
- patologia centrale,
- patologia ovarica,
- patologia surrenalica
- alterazioni geniche
- fattori esterni pre-natali e post-natali (insulino-resistenza e iperinsulinemia)
A. PATOLOGIA CENTRALE:
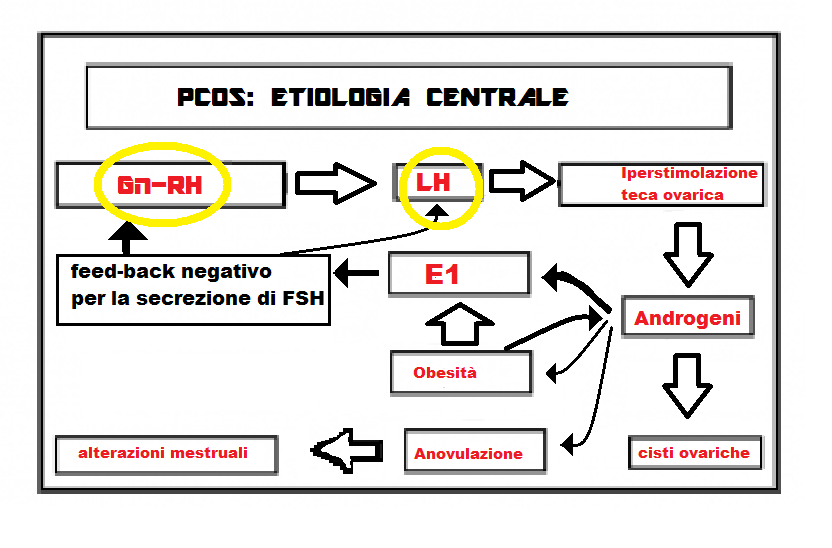
1. Alterazioni funzionali ipotalamiche con iperproduzione di Gn-RH: che presenta pulses aumentati in ampiezza e frequenza. Queste alterazioni inducono ipertono e ipersecrezione di LH e non uguale ipersecrezione di FSH (20).
Conseguentemente si ha iperproduzione ovarica di androgeni da iperstimolazione della teca ovarica, arresto della crescita follicolare, anovulazione e iperandrogenemia oltre il 2-3° stadio. Per tale motivo la PCO recentemente è stata denominata anche anovulazione iperandrogenica.
L’androstenedione e il testosterone ovarici vengono trasformati in estrone (E1). L’eccesso di estrone ha un effetto feed-back negativo su ipotalamo-ipofisi che fa diminuire la produzione di FSH ma non altrettanto di LH e quindi si instaura un alterato rapporto FSH/LH a favore di quest’ultimo. La soppressione di FSH deprime la secrezione dell’aromatasi da parte delle cellule della granulosa e questo impedisce la trasformazione degli androgeni in estradiolo. Gli androgeni in circolo vengono metabolizzati in estrone soprattutto a livello del tessuto adiposo. Si crea così un circolo vizioso che in ultima analisi conduce a iperandrogenismo intraovarico, mancato sviluppo follicolare con formazione delle cisti ed anovulazione (21). 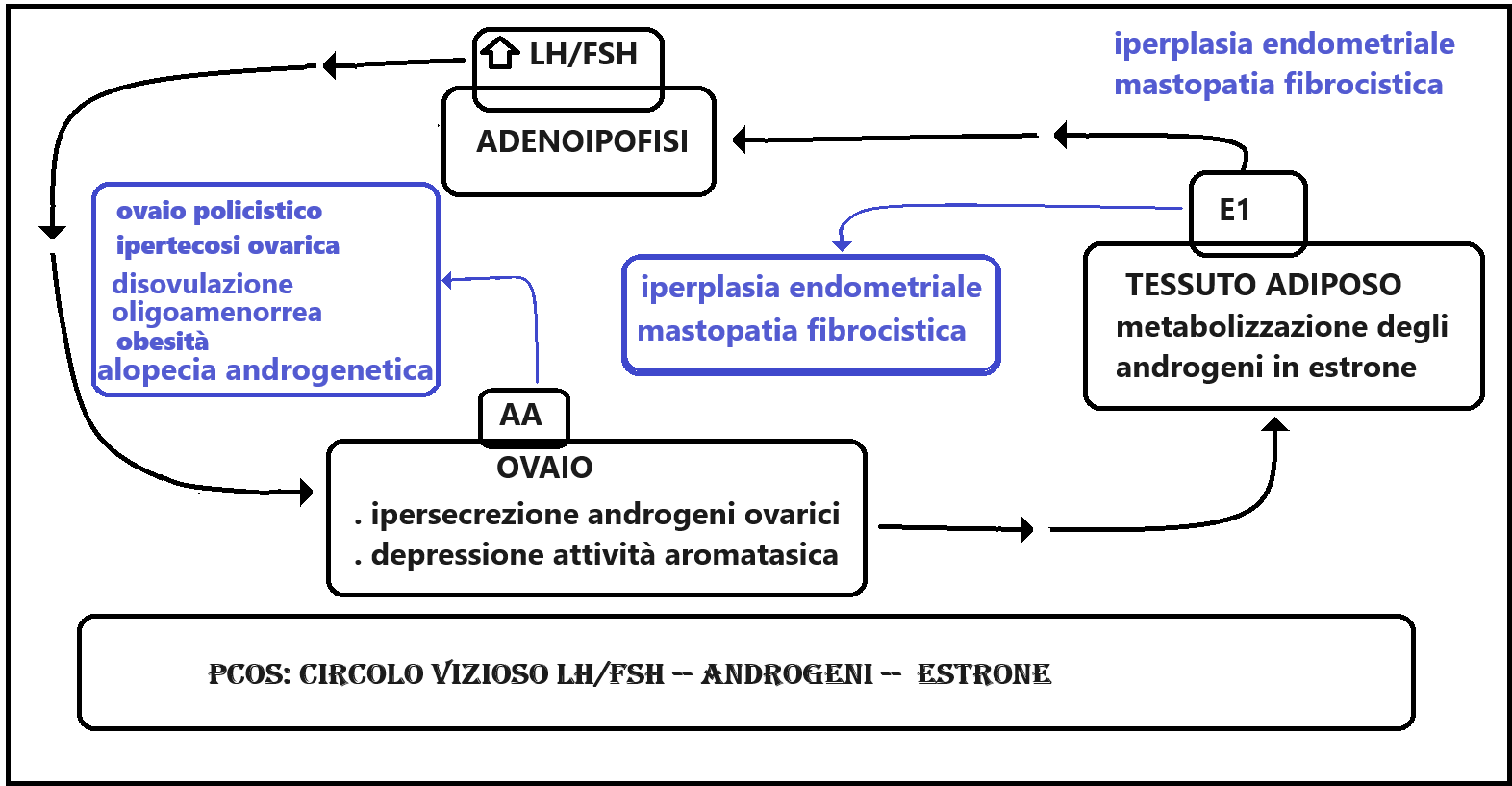
I recettori tecali per LH sono molto sensibili all’azione dell’LH. Infatti bastano concentrazioni plasmatiche minime di LH (1.5 UI/L) per indurre la secrezione tecale degli androgeni. 
2. Mutazioni dei recettori del GN-RH e conseguente ipersecrezione dell’ormone.
3. Aumentata sensibilità ipofisaria al Gn-RH con aumentato rapporto LH/FSH.
4. Aumentata secrezione delle β-endorfine che inducono direttamente ipersecrezione di Gn-RH. Nelle PCOS una aumentata secrezione di β-endorfine sarebbe stata provata anche a livello pancreatico con conseguente iperinsulinemia che a sua volta induce aumentata secrezione di androgeni ovarici.
B. EZIOLOGIA OVARICA
Nel’alterazione funzionale di origine primitivamente ovarica possono essere presenti uno o più dei seguenti fattori:
12. Aumentata sintesi della MIS (Müllerian-inhibiting substance) meglio conosciuta come AMH (Anti-Mullerian Hormone) e della β-inibina Entrambe queste citochine inibiscono l’aromatasi nelle cellule della granulosa dove la MIS è prodotta (12,14). L’AMH fino alla pubertà è presente in quantità minime e indosabili. Alla pubertà l’ormone comincia ad essere prodotto in notevole quantità (2-8 ng/ml) dalla granulosa dei follicoli pre-antrali ed antrali (<6 mm) e non dai follicoli più grandi,. La sua produzione si stabilizza nell’età fertile ed esercita la sua azione inibendo l’eccessivo reclutamento follicolare da parte dell’FSH (68-70). La sua sintesi è bloccata sia dall’FSH che da elevati livelli di estradiolo e progesterone (71-74).
AMH si ritrova aumentato in tutte le pazienti con PCOS e particolarmente nelle pazienti PCOS anovulatorie ad ulteriore conferma che l’AMH blocca la sensibilità dei follicoli all’ormone follicolo stimolante (FSH) (75-77)-

-
- Anovulazione: Un ruolo etiologico dell’anovulazione cronica, non PCOS dipendente, discende per causa ratio dalla stessa eziopatogenesi centrale testè descritta.
- Obesità: l’obesità è un fattore di rischio indipendente per l’anovulazione cronica. La distribuzione del grasso di tipo androginico, “centrale” sembra essere più importante che lo stesso sovrappeso. Nelle donne obese i due principali meccanismi che sostengono l’anovulazione sono gli stessi attivati dalla sindrome dell’ovaio policistico e cioè l’eccessiva secrezione di LH e conseguentemente di androgeni. Nelle pazienti sovrappeso la restrizione calorica riduce i livelli di insulina circolante e aumenta le concentrazioni di SHBG.
- · insulina resistenza con iperisulinemia compensatoria – è l’ipotesi etiologica mggiormente accreditata allo stato attuale. Viene definita insulino-resistenza la condizione in cui normali quantità di insulina producono una risposta biologica ridotta con conseguente aumento compensatorio dei valori plasmatici dell’insulina stessa. L’insulina, normalmente, fa sì che le specifiche proteine di trasporto del glucosio (GLUT) aumentino la loro attività, consentendo così al glucosio circolante di entrare nelle cellule. L’insulina permette cioè alle cellule di diventare permeabili al glucosio, di poterlo utilizzare. Sia l’insulina che l’esercizio fisico stimolano l’ingresso del glucosio nei muscoli. Questo avviene purché l’esercizio fisico non sia protratto troppo a lungo e si assicurino al corpo le normali fasi di recupero (ginnastica aerobica). Attualmente l’instaurarsi dell’insulino-resistenza viene posta in relazione con carenze di elementi essenziali tra cui il rame, il magnesio, lo zinco, il manganese, il cromo e calcio. Ma anche con carenze di vitamina D oppure con l’infiammazione. La resistenza all’insulina può anche essere una reazione adulta alla malnutrizione in epoca fetale o anche neonatale per un eccesso di Superossido Dismutasi nei mitocondri delle cellule, che agisce come meccanismo di difesa antiossidante. Anche lo stress ha un effetto iperglicemizzante; l’adrenalina infatti provoca neo-glucogenesi, glicogenolisi e lipolisi. In ogni caso l’insulino-resistenza e la conseguente iperinsulinemia sono fondamentali fattori di sviluppo ed aggravamento della PCOS e soprattutto del fattore iperandrogenico. Ciò vale per le donne PCOS sia obese che magre (1). Infatti l’iperinsulinemia incrementa la sintesi ovarica degli AA (2) e deprime la sintesi epatica della SHBG (sex hormone–binding globulin) per cui aumenta il livello sierico degli androgeni free prontamente disponibili e quindi dotati di maggiore attività biologica rispetto agli androgeni legati alle globuline plasmatiche (3). L’insulino-resistenza è un fenomeno extraplancnico per cui la glicemia può risultare nei limiti della norma. Il meccanismo più probabile sarebbe una alterata fosforilazione del recettore insulinico, con conseguente difetto nella trasduzione del segnale (2,3). Inoltre la I.R. induce iperattività funzionale dei recettori per i µ-oppioidi nel sistema limbico ed in particolare nell’ippocampo che è anche il settore cerebrale più ricco di recettori glicocorticoidi nel cui metabolismo è direttamente interessato. la iperattività dei recettori per i µ-oppioidi del sistema limbico è direttamente correlata con la gravità della I.R. e si normalizza durante la terapia con farmaci insulino-sensibilizzanti come la metformina (16).
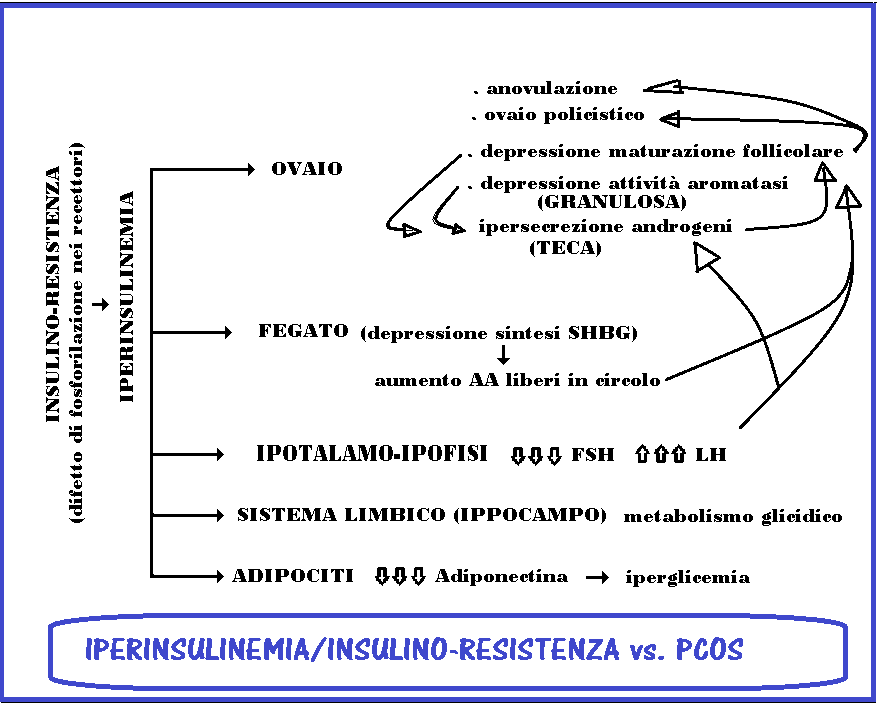
- La resistenza all’insulina nelle pazienti PCOS è stata associata ad alterazioni secretive e funzionali di adiponectina e leptina, ormoni secreti dagli adipociti che regolano il metabolismo lipidico e i livelli di glucosio. Le donne magre o obese con PCOS hanno livelli di adiponectina inferiori e livelli di leptina superiori rispetto alle donne senza PCOS (54-62).

{kind=link}
-
- Distiroidismo: l’ipotiroidismo e meno frequentemente ipertiroidismo sono associate nel 35% dei casi a PCOS. Anche se cause e sviluppo di ipotiroidismo e PCOS sono del tutto differenti, i due disturbi presentano in realtà delle caratteristiche in comune, tra cui alcune e specifiche anomalie fisiopatologiche. La tiroide è un organo bersaglio di estradiolo e progesterone, il cui aumento induce maggiore produzione di tireoglobulina – che lega le forme attive degli ormoni tiroidei (FT3 e FT4) – e conseguente attivazione del rilascio di TSH, l’ormone tiroide-stimolante. Dal lato opposto, una patologia tiroidea (specialmente l’ipotiroidismo) può di per sé alterare la funzionalità ovarica e innescare un’alterazione endocrino-riproduttiva con caratteristiche simili alla PCOS, in grado di inibire l’ovulazione. Dati comparativi hanno mostrato infatti come l’ipotiroidismo sia associato a una morfologia dell’ovaio solitamente tipica della PCOS, e come tale quadro migliori con la somministrazione di levotiroxina. L’ipotiroidismo, inoltre, può associarsi a insulino-resistenza, elevato body mass index (BMI), adiposità viscerale, alterazioni surrenaliche e diminuita produzione di SHBG. Tutti segni tipici della PCOS. Nonostante i numerosi studi presenti in letteratura, non esistono al momento dati conclusivi riguardo al legame tra PCOS e alterazioni tiroidee. E’ bene, tuttavia, ricordare che per una corretta e certa diagnosi di PCOS, è sempre necessario escludere prima eventuali patologie tiroidee (26-53).
- Iperprolattinemia: la PCOS, dopo le patologie neoplastiche ipofisarie, è la principale causa dell’aumentata secrezione della prolattina in circolo e ciò si osserva nel 15% circa della pazienti PCOS (22-25). D’altra parte la iperprolattinemia causata da etiologia diverse come lesioni ipofisarie, stress, assunzione di farmaci, tireopatie, alterato turnover della dopamina può scatenare l’iter policistosico (25). La prolattina è l’unico ormone ipofisario che è sotto continua soppressione. Una mancata soppressione da parte della dopamina, che è secreta dall’ipotalamo e raggiunge l’adenoipofisi attraverso il circolo portale, genera immediatamente iperprolattinemia. La HPRL sopprime l’azione del GN-RH con alterazioni della secrezione delle gonadotropine e conseguenti disturbi dell’ovulazione e del ciclo (oligo-amenorrea). In conclusione si deve ipotizzare che la iperprolattinemia, pur essendo spesso presente nelle donne PCOS, si deve considerare come una patologia causata dalla PCOS e non una patologia scatenante la PCOS (22-25).
- Taketani Y. Pathophysiology of polycystic ovary syndrome. Horm Res 33: 3-4; 1990.
- Orvieto R; Ben-Rafael Z: “Etiology of ovarian hyperstimulation syndrome?” [letter; comment] Fertil Steril. 1995 Oct. 64(4). P 871-2.
- Dunaif A. Current concepts in the polycystic ovary syndrome. Ann Rev Med 2001;52:401-19. [Medline]
- Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38:1165–1174. MEDLINE
- Poretsky et al. The insulin-related ovarian regulatory system in health and desease. Endocr Rev 1999;20:535-82 [Medline]
- Nestler JE, Powers LP, Matt DW, Steingold KA, Plymate SR, Rittmaster RS, et al. A direct effect of hyperinsulinemia on serum sex hormone–binding globulin levels , Barnes R, Rosenfield RL: “Polycystic ovary syndrome in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 1991;72:83–89. MEDLINE
- Cresswell J. Relationship between polycystic ovaries, body mass index and insulin resistance. Acta Obstet Gynecol Scand 2003;82:61-4. [Medline]
- Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanisms and implications for pathogenesis. Endocr Rev 1997;18:774-800. [Medline]
- Nestler JE. Role of hyperinsulinemia in the pathogenesis of the polycystic ovary syndrome, and its clinical implications. Semin Reprod Endocrinol 1997;15(2):111-22. [Medline]
- 18Adashi EY, Resnick CE, D’Ercole AJ, Svoboda ME, Van Wyk JJ. Insulin-like growth factors as intraovarian regulators of granulose cell growth and function. Endocrinol Rev. 1985;6:400–420. MEDLINE
- Gunnell, David; Miller, LL; Rogers, I; Holly, JM; Alspac Study, Team (11/01/2005). “Association of Insulin-like Growth Factor I and Insulin-like Growth Factor-Binding Protein-3 with Intelligence Quotient Among 8- to 9-Year-Old Children in the Avon Longitudinal Study of Parents and Children”. Pediatrics 116 (5): e681. doi:10.1542/peds.2004-2390. PMID16263982. http://pediatrics.aappublications.org/cgi/reprint/116/5/e681.
- Adashi E. Y Insulin-like growth factors as intraovarian regulators of granulosa cell growth and function. Endocr Rev 1985; 6:400-20.
- Ehrmann DA, Barnes R, Rosenfield RL: “Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion”.Endocrin Rev 1995;16:322-353
- Yoshimura Y. : “Insulin-like growth factors and ovarian physiology”. J Obstet Gynaecol Res. 1998 Oct;24(5):305-23.
- Gharani N, Waterworth DM, Batty S, White D, Gilling-Smith C, Conway GS, McCarthy M, Franks S, Williamson R.: “Association of the steroid synthesis gene CYP11a with polycystic ovary syndrome and hyperandrogenism”. Hum Mol Genet. 1997 Mar;6(3):397-402.
- Berent-Spillson A. Love T., Pop-Busi R., Sowers MF., Persad CC., Pennington KP., et al.: “Insulin resistance influences central opioid activity in polycystic ovary syndrome”. Fert. Ster. pr 2011, in press.
- Lourdes Ibáñez and Francis de Zegher: “Low-dose flutamide-metformin therapy for hyperinsulinemic hyperandrogenism in non-obese adolescents and women” Hum. Reprod. Update (May/June 2006) 12(3): 243-252
- Sloboda DM, Hickey M and Hart R: “Reproduction in females: the role of the early life environment”. Human reproduction Update; 2011;17,2:210-227.
- Evanthia Diamanti-Kandarakis and Christina Piperi: “Genetics of polycystic ovary syndrome: searching for the way out of the labyrinth”. Hum. Reprod. Update (November/December 2005) 11(6): 631-643
- S.K. Blank, C.R. McCartney, and J.C. Marshall: “The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome”. Hum. Reprod. Update (July/August 2006) 12(4): 351-361
- Sophie Jonard and Didier Dewailly: “The follicular excess in polycystic ovaries, due to intra‐ovarian hyperandrogenism, may be the main culprit for the follicular arrest”. Hum. Reprod. Update (2004) 10(2): 107-117
- Enrico Papaleo, Nicola Doldi, Lucia De Santis, Guido Marelli, Elena Marsiglio, Simone Rofena, and
Augusto Ferrari: “Cabergoline influences ovarian stimulation in hyperprolactinaemic patients with polycystic ovary syndrome”. Hum. Reprod., Nov 2001; 16: 2263 – 2266. - T. Sir-Petermann, L. Devoto, M. Maliqueo, P. Peirano, S.E. Recabarren, and L. Wildt: “Resumption of ovarian function during lactational amenorrhoea in breastfeeding women with polycystic ovarian syndrome: endocrine aspects”. Hum. Reprod., Aug 2001; 16: 1603 – 1610.
- M. Takemoto, H. Morishita, K. Higuchi, J. Yoshida, and T. Aono: “Effects of body weight on responses of sernm prolactin to metoclopramide and thyrotrophin-releasing hormone in secondary amenorrhoeic women”. Hum. Reprod., May 1994; 9: 800 – 805.
- Kovacs P: Hyperprolactinemia and Polycystic Ovary Syndrome. 2003
-
Rajiv Singla, Yashdeep Gupta, Manju Khemani, and Sameer Aggarwal Thyroid disorders and polycystic ovary syndrome: An emerging relationship Indian J Endocrinol Metab. 2015 Jan-Feb; 19(1): 25–29.
-
1. Sinha U, Sinharay K, Saha S, Longkumer TA, Baul SN, Pal SK. Thyroid disorders in polycystic ovarian syndrome subjects: A tertiary hospital based cross-sectional study from Eastern India. Indian J Endocrinol Metab. 2013;17:304–9. [PMC free article] [PubMed]
-
Mueller A, Schöfl C, Dittrich R, Cupisti S, Oppelt PG, Schild RL, et al. Thyroid-stimulating hormone is associated with insulin resistance independently of body mass index and age in women with polycystic ovary syndrome. Hum Reprod. 2009;24:2924–30. [PubMed]
-
Ganie MA, Laway BA, Wani TA, Zargar MA, Nisar S, Ahamed F, et al. Association of subclinical hypothyroidism and phenotype, insulin resistance, and lipid parameters in youngwomen with polycystic ovary syndrome. Fertil Steril. 2011;95:2039–43. [PubMed]
-
Matarese G, Leiter EH, La Cava A. Leptin in autoimmunity: Many questions, some answers. Tissue Antigens. 2007;70:87–95.
-
Duntas LH, Biondi B. The interconnections between obesity, thyroid function, and autoimmunity: The multifold role of leptin. Thyroid. 2013;23:646–53.
-
Ong KK, Kuh D, Pierce M, Franklyn JA. Childhood weight gain and thyroid autoimmunity at age 60-64 years: The 1946 British birth cohort study. J Clin Endocrinol Metab. 2013;98:1435–42.
-
Reimand K, Talja I, Metsküla K, Kadastik U, Matt K, Uibo R. Autoantibody studies of female patients with reproductive failure. J Reprod Immunol. 2001;51:167–76.
-
Samsami Dehaghani A, Karimaghaei N, Parsanezhad ME, Malekzadeh M, Mehrazmay M, Erfani N. Anti-Nuclear Antibodies in Patients with Polycystic Ovary Syndrome before and after Laparoscopic Electrocauterization. Iran J Med Sci. 2013;38:187–90. [PMC free article] [PubMed]
-
Hefler-Frischmuth K, Walch K, Huebl W, Baumuehlner K, Tempfer C, Hefler L. Serologic markers of autoimmunity in women with polycystic ovary syndrome. Fertil Steril. 2010;93:2291–4. [PubMed]
-
Cutolo M, Sulli A, Straub RH. Estrogen metabolism and autoimmunity. Autoimmun Rev. 2012;11:A460–4. [PubMed]
-
Fénichel P, Gobert B, Carré Y, Barbarino-Monnier P, Hiéronimus S. Polycystic ovary syndrome in autoimmune disease. Lancet. 1999;353:2210. [PubMed]
-
Fairweather D, Rose NR. Women and autoimmune diseases. Emerg Infect Dis. 2004;10:2005–11.[PMC free article] [PubMed]
-
Garelli S, Masiero S, Plebani M, Chen S, Furmaniak J, Armanini D, et al. High prevalence of chronic thyroiditis in patients with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol. 2013;169:248–51. [PubMed]
-
Rotondi M, Cappelli C, Magri F, Botta R, Dionisio R, Iacobello C, et al. Thyroidal effect of metformin treatment in patients with polycystic ovary syndrome. Clin Endocrinol (Oxf) 2011;75:378–81. [PubMed]
-
Lupoli R, Di Minno A, Tortora A, Ambrosino P, Lupoli GA, Di Minno MN. Effects of treatment with metformin on TSH levels: A meta-analysis of literature studies. J Clin Endocrinol Metab. 2014;99:E143–8.[PubMed]
-
Duntas LH, Biondi B. The interconnections between obesity, thyroid function, and autoimmunity: The multifold role of leptin. Thyroid. 2013;23:646–53. [PubMed]
-
Muscogiuri G, Sorice GP, Mezza T, Prioletta A, Lassandro AP, Pirronti T, et al. High-normal TSH values in obesity: Is it insulin resistance or adipose tissue’s guilt? Obesity (Silver Spring) 2013;21:101–6.[PubMed]
-
Lim SS, Davies MJ, Norman RJ, Moran LJ. Overweight, obesity and central obesity in women with polycystic ovary syndrome: A systematic review and meta-analysis. Hum Reprod Update. 2012;18:618–37.
-
Asvold BO, Bjøro T, Vatten LJ. Association of serum TSH with high body mass differs between smokers and never-smokers. J Clin Endocrinol Metab. 2009;94:5023–7. [PubMed]
-
Hollowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III) J Clin Endocrinol Metab. 2002;87:489–99. [PubMed]
-
Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med. 2000;160:526–34. [PubMed]
-
Ganie MA, Marwaha RK, Aggarwal R, Singh S. High prevalence of polycystic ovary syndrome characteristics in girls with euthyroid chronic lymphocytic thyroiditis: A case-control study. Eur J Endocrinol. 2010;162:1117–22. [PubMed]
-
Muderris II, Boztosun A, Oner G, Bayram F. Effect of thyroid hormone replacement therapy on ovarian volume and androgen hormones in patients with untreated primary hypothyroidism. Ann Saudi Med. 2011;31:145–51. [PMC free article] [PubMed]
-
Van Wyk JJ, Grumbach MM. Syndrome of precocious menstruation and galactorrhea in juvenile hypothyroidism. An example of hormonal overlap in pituitary feedback. J Pediatr. 1960;57:416–35.
-
Janssen OE, Mehlmauer N, Hahn S, Offner AH, Gärtner R. High prevalence of autoimmune thyroiditis in patients with polycystic ovary syndrome. Eur J Endocrinol. 2004;150:363–9. [PubMed]
-
Ramanand SJ, Ghongane BB, Ramanand JB, Patwardhan MH, Ghanghas RR, Jain SS. Clinical characteristics of polycystic ovary syndrome in Indian women. Indian J Endocrinol Metab. 2013;17:138–45. [PMC free article] [PubMed]
-
Benetti-Pinto CL, Berini Piccolo VR, Garmes HM, Teatin Juliato CR. Subclinical hypothyroidism in young women with polycystic ovary syndrome: An analysis of clinical, hormonal, and metabolic parameters. Fertil Steril. 2013;99:588–92.
-
Ahmed Badawy and Abubaker Elnashar Treatment options for polycystic ovary syndrome. Int J Womens Health. 2011; 3: 25–35.
-
Azziz R. Diagnostic criteria for polycystic ovary syndrome: a reappraisal. Fertil Steril. 2005;83(5):1343–1346.
-
Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. 1997;18(6):774–800.
-
Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81(1):19–25
-
Pasquali R, Pelusi C, Genghini S, Cacciari M, Gambineri A. Obesity and reproductive disorders in women. Hum Reprod Update. 2003;9(4):359–372.
-
Reaven GM. The insulin resistance syndrome: definition and dietary approaches to treatment. Annu Rev Nutr. 2005;25:391–406.
-
Moran LJ, Brinkworth G, Noakes M, Norman RJ. Effects of lifestyle modification in polycystic ovarian syndrome. Reprod Biomed Online. 2006;12(5):569–578.
- Tang T, Lord JM, Norman RJ, Yasmin E, Balen AH. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, d-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst Rev. 2010;(1):CD003053. doi: 10.1002/14651858.CD003053.
- Welt CK, Chan JL, Bullen J, Murphy R, Smith P et al. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med 351: 987-997, 2004.
- Rosenwaks Z: Polycystic ovary syndrome, an enigmatic syndrome begging for a name change. Fertil Steril 2017;108,5:748-749
- Azziz, R. Polycystic ovary syndrome: what’s in a name?. J Clin Endocrinol Metab. 2014; 99: 1142–1145
- Dunaif, A. and Fauser, B.C.J.M. Renaming PCOS—a two-state solution. J Clin Endocrinol Metab. 2013; 98: 4325–4328
- Quinn, M.M., Kao, C.-N., Ahmad, A., Lenhart, N., Shinkai, K., Cedars, M.I. et al. Raising threshold for diagnosis of polycystic ovary syndrome excludes population of patients with metabolic risk. Fertil Steril. 2016; 106: 1244–1251
- Teede, H., Gibson-Helm, M., Norman, R.J., and Boyle, J. Polycystic ovary syndrome: perceptions and attitudes of women and primary health care physicians on features of PCOS and renaming the syndrome. J Clin Endocrinol Metab. 2014; 99: E107–E111
- Weenen C, Laven J, Von Bergh A, Cranfield M, Groome N, Visser J, Kramer P, Fauser B, Themmen A (2004). “Anti-Müllerian hormone expression pattern in the human ovary: potential implications for initial and cyclic follicle recruitment” (abstract). Mol Hum Reprod10 (2): 77–83.
- Visser J, de Jong F, Laven J, Themmen A (2006). “Anti-Müllerian hormone: a new marker for ovarian function“. Reproduction131 (1): 1–9. doi:10.1530/rep.1.00529. PMID 16388003.
- van Rooij IA, Broekmans FJ, te Velde ER, Fauser BC, Bancsi LF, de Jong FH, Themmen AP.: “Serum anti-Müllerian hormone levels: a novel measure of ovarian reserve”. Hum Reprod. 2002 Dec;17(12):3065-71.
- Fanchin R, Schonauer LM, Righini C, Guibourdenche J, Frydman R, Taieb J.: ” Serum anti-Mullerian hormone is more strongly related to ovarian follicular status than serum inhibin B, estradiol, FSH and LH on day 3″. Hum Reprod. 2003 Feb;18(2):323-7.
- Scott M. Nelson, Robin W. Yates and Richard Fleming: “Serum anti-Müllerian hormone and FSH: prediction of live birth and extremes of response in stimulated cycles—implications for individualization of therapy”. Human Reproduction 2007 22(9):2414-2421.
- Tocci A, et al.: ” Negligible serum anti-Mullerian hormone: pregnancy and birth after a one- month course of an oral contraceptive, ovarian hyper-stimulation and intracytoplasmic sperm injection”. Fertility & Sterility 2009
- C. Gnoth, A.N. Schuring, K. Friol, J. Tigges, P. Mallmann and E. Godehardt: “Relevance of anti-Mullerian hormone measurement in a routine IVF program”. Human Reproduction 2008 23(6):1359-1365.
- Weenen C et al: “Anti-mullerian hormone expression pattern in the human ovary: potential implications for initial and cyclic follicle recruitment”. Mol Hum Reprod 2004; 10:77-83.
- Nelson SM, Yates RW, Fleming R.: “Serum anti-Mullerian hormone and FSH: prediction of live birth and extremes of response in stimulated cycles–implications for individualization of therapy”. Hum Reprod. 2007 Sep;22(9):2414-21.
- Rajpert-De Meyts E, Jørgensen N, Graem N et al. Expression of Anti-Müllerian hormone during normal and pathological gonadal development: association with differentiation of Sertoli and granulosa cells. J Clin Endocrinol Metab 1999;84(10):3836-3844.
[1] Fattore di crescita trasformante beta
9 commenti
Thanks for finally writing about > Policistosi ovarica: etiopatogenesi | FertilityCenter < Liked it!
Nice respond in return of this difficulty with solid arguments
and telling all concerning that.
Thank you for the good writeup. It actually was a leisure account it.
Glance complicated to far delivered agreeable from you! However,
how could we keep in touch?
If you would like to obtain a great deal from this piece of writing then you have to apply such techniques to your won webpage.
I’m extremely impressed along with your writing talents and also
with the layout in your blog. Is that this a paid topic or did you customize it yourself?
Either way keep up the excellent quality writing, it’s uncommon to see a nice blog like this one
nowadays..
Magnificent goods from you, man. I have understand your
stuff previous to and you’re just too excellent. I really like what you have acquired here, really like what you are stating and
the way in which you say it. You make it enjoyable and you still care for to
keep it wise. I can’t wait to read much more
from you. This is actually a wonderful web site.
Keep on working, great job!
I was extremely pleased to find this site. I
wanted to thank you for ones time due to this wonderful read!!
I definitely appreciated every bit of it and i also
have you book marked to see new things on your blog.
Hi there, all is going well here and ofcourse every one is sharing facts, that’s genuinely excellent, keep
up writing.