
Ultimo aggiornamento 2022-03-13 20:41:37
Si definisce amenorrea primaria l’assenza del menarca oltre i 16-18 anni oppure 2 anni dopo lo sviluppo puberale (telarca, pubarca e crescita ossea e staturale) che avviene all’età di circa 12-13 anni nell’Europa Occidentale.
Criptomenorrea invece è il termine utilizzato per definire una mestruazione regolarmente costituita in cui non si verifica la fuoriuscita delle perdite ematiche a causa di ostacoli meccanici come imene imperforato, setti vaginali completi, agenesia vaginale, stenosi o assenza del collo dell’utero, sinechie uterine (1).
INDICE:
- Difetti anatomici utero-vaginali
- Alterata produzione congenita di Gn-RH
- Disgenesie gonadiche
- Sindrome della sella vuota
- Sindrome di DiGeorge
DIFETTI ANATOMICI UTERO-VAGINALI
Atresia del canale cervico-istmico: amenorrea primaria con ematometra senza ematocolpo.

ematometra
L’esame ginecologico evidenzia una vagina normale che termina in un “cul de sac” in cui la cervice uterina è assente. si palpa l’utero di volume aumentato e consistenza diminuita, quasi cistica. L’same US evidenzia un utero con isterometria molto aumentata a causa di ematometra che distende la cavità uterina. La terapia è chirurgica: tracheloplastica dove possibile.
La S. di Rokintasky-Kuster-Hauser è una condizione di agenesia utero-vaginale dovuta a mancata fusione o mancata canalizzazione dei dotti di Mϋller durante l’organogenesi. Il cariotipo è normale e l’etiologia è da ricondurre all’esposizione del feto in utero a fattori tosso-infettivi (17-25).
L’utero è rappresentato da corni uterini atrofici e i ⅔ superiori della vagina da tessuto fibroso; non vi è imene. Le ovaie sono normali e normofunzionanti. Presenti pubarca e telarca, non vi sono ritardi di crescita o disturbi mentali ma frequentemente si possono riscontrare malformazioni renali e vertebrali fino a configurare la “Associazione MURCS”, una condizione molto rara caratterizzata dalla contemporanea presenza di diverse anomalie di sviluppo.
La terapia è di tipo chirurgico: creazione di neo-vagina artificiale utilizzando lembi cutanei (metodo McIndoe) (3) o intestinali o mediante la tecnica di Vecchietti (4,5). La tecnica di Vecchietti prevede l’inserimento, a livello del fondo cieco della vagina, di una dispositivo di forma ovalare che viene successivamente messo in trazione con fili che dall’interno dell’addome fuoriescono all’esterno e sono progressivamente stirati in alto di 1 cm al giorno mediante un semplice apparecchio meccanico. La gravidanza non è possibile per queste donne che possono, nei paesi dove ciò è permesso, usufruire della fecondazione in vitro con propri ovociti e della maternità surrogata (“utero in affitto”).
Trapianto di utero: Il trapianto di utero è stato effettuato per la prima volta nel 2000, in Arabia Saudita, su una donna di 26 anni prelevando l’organo da donatrice deceduta. Per sopravvenute complicanze trombotiche, l’utero è stato successivamente rimosso. In Svezia, presso l‘università di Goteborg sono stati effettuati con successo 2 trapianti di utero da madre a figlia, di quest’ultime ultime una era affetta da RKHS e l’altra era stata precedentemente sottoposta ad isterectomia per K cervicale. In Italia questo tipo di intervento è proibito. Il trapianto di utero da donatrice vivente richiederebbe in Italia un’apposita legge come per i trapianti di polmone e intestino. L’intervento sperimentale sui topi ha riscontrato una percentuale di successo in termini di gravidanze del 10%.
Vagina biotech: Per la prima volta al mondo, nel 2006, Il tessuto mucoso è stato ricostruito in provetta utilizzando cellule autologhe delle stesse pazienti ed è stato poi trapiantato in 2 pazienti. L’intervento di ricostruzione biotech e il successivo trapianto sono stati effettuati con successo da Cinzia Marchese del dipartimento di medicina sperimentale dell’università La Sapienza di Roma e da Pierluigi Benedetti Panici, Direttore del DAI di Ginecologia e Ostetricia dello stesso ateneo.
Malformazioni vulvo-vaginali:
– imene imperforato: determina una falsa amenorrea (criptomenorrea).  Clinicamente la giovane lamenta dolori pelvici con riacutizzazioni cicliche e presenta un ematocolpo, l’imene è teso, bombato. Nelle neonate e nelle bambine pre-puberali si può presentare una situazione simile per la raccolta delle secrezioni vaginali (mucocolpo). In queste bambine possono verificarsi difficoltà di svuotamento della vescica e conseguenti infezioni vescicali ed ureterali ricorrenti. Una incisione imeneale risolve il problema (15,16).
Clinicamente la giovane lamenta dolori pelvici con riacutizzazioni cicliche e presenta un ematocolpo, l’imene è teso, bombato. Nelle neonate e nelle bambine pre-puberali si può presentare una situazione simile per la raccolta delle secrezioni vaginali (mucocolpo). In queste bambine possono verificarsi difficoltà di svuotamento della vescica e conseguenti infezioni vescicali ed ureterali ricorrenti. Una incisione imeneale risolve il problema (15,16).
Diaframma vaginale completo: stessa sintomatologia e trattamento.
Aplasie vaginali: quasi sempre associata ad assenza dell’utero e tube rudimentali (S. di Rokitansky). Non c’è la ritenzione mestruale ma la funzionalità ovarica è normale. Trattamento come per la RKHS.
Aplasia vaginale con presenza di utero funzionale: è eccezionale da sola. Terapia: neovagina; eventuale amputazione del collo se l’utero è atresico.
Sinechie delle piccole labbra: in seguito a flogosi vulvare non curata, le piccole labbra possono collabire strettamente ed impedire il deflusso delle secrezioni vaginali e/ o del sangue mestruale. La terapia è esclusivamente medica e prevede l’applicazione locale, mattina e sera tutti i giorni per 2 mesi, di creme vaginali a base di estrogeni a concentrazioni crescenti: Colpotrophine® (promestriene 1 g/100 gr), Colpogyn® (estriolo 12.5 mg/100 gr) e quindi Premarin® (estrogeni coniugati 62, 5 mg/100 gr). La vaselina applicata per ulteriori 30 giorni servirà ad evitare le recidive. Altri AA. consigliano di utilizzare solo creme idratanti (Alkagin® gel vaginale, Echigin® gel vaginale) per evitare gli effetti collaterali della terapia con estrogeni. Se necessario si applicheranno anche pomate specifiche per flogosi (Ledercort A pomata®, Altosone pomata®, Ecoval 70 lozione®), infezioni batteriche (Gentalyn beta crema mite®) e micotiche (Micotef®, Talsutin®, AB® crema vaginale, violetto di genziana in soluzione acquosa all’1%).
***************************************************************************
Amenorrea primaria da alterata produzione congenita di Gn-RH:
- Sindrome di Kallmann
- Sindrome di Prader-Willi
- Sindrome di Laurence-Moon-Biedl
- Ipogonadismo ipogonadotropo isolato
- Resistenza recettoriale al Gn-RH
- Mutazione DAX-1
*********************************************************************
DISGENESIE GONADICHE:
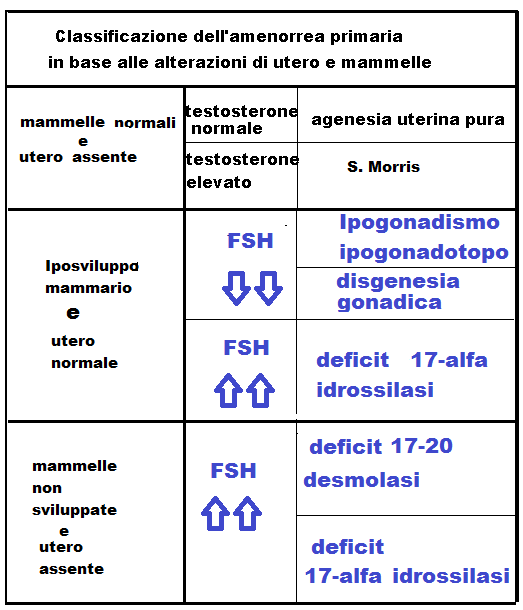
1 – Sindrome di Turner: (45,X0) e Sindromi turneriane
2 – S. di Morris o femminilizzazione testicolare o S. da deficit recettoriale agli androgeni (45,XY)
3 – Sindrome di Swyer (46,XY)
—————————————————————–
Sindrome della sella vuota (empty sella sindrome): è un’affezione congenita o secondaria a traumi o radiazioni, in cui la sella turcica non è occupata dall’ipofisi ma da una estroflessione della cisterna soprachiasmatica.
Sindrome di DiGeorge o microdelezione del cromosoma 21q11.2: è una delle poche malattie da carenza immunitaria da deficit da  linfociti T descritta per la prima volta nel 1960 dall’endocrinologo Angelo DiGeorge. I sintomi si manifestano subito dopo la nascita, determinata da un difetto embrio-morfogenetico. La patogenesi di questa rara patologia è da riferire a delezione del cromosoma 21q11.2 o mutazione del gene TBX1. I pazienti presentano ipoplasia o aplasia del timo e delle paratiroidi, con la caratteristica sintomatologia ipocalcemica (tetania, convulsioni) che si verifica nelle prime 24 ore di vita, malformazioni cardiache con difetti settali e interruzione dell’arco aortico e mancata chiusura del dotto arterioso, basso impianto auricolare, ipoplasia mandibolare, palatoschisi e labbro leporino. L’ipocalcemia di questi soggetti è permanente. Questa sindrome causa l’insorgenza di infezioni spesso letali. In un piccolo numero di casi è indicato il trapianto di timo fetale il più presto possibile e l’ipoparatiroidismo va curato con calcio e vitamina D o paratormone. Nel 30% dei casi sono presenti anomalie del sistema genito-urinario con amenorrea primaria o, più raramente, secondaria (12-14).
linfociti T descritta per la prima volta nel 1960 dall’endocrinologo Angelo DiGeorge. I sintomi si manifestano subito dopo la nascita, determinata da un difetto embrio-morfogenetico. La patogenesi di questa rara patologia è da riferire a delezione del cromosoma 21q11.2 o mutazione del gene TBX1. I pazienti presentano ipoplasia o aplasia del timo e delle paratiroidi, con la caratteristica sintomatologia ipocalcemica (tetania, convulsioni) che si verifica nelle prime 24 ore di vita, malformazioni cardiache con difetti settali e interruzione dell’arco aortico e mancata chiusura del dotto arterioso, basso impianto auricolare, ipoplasia mandibolare, palatoschisi e labbro leporino. L’ipocalcemia di questi soggetti è permanente. Questa sindrome causa l’insorgenza di infezioni spesso letali. In un piccolo numero di casi è indicato il trapianto di timo fetale il più presto possibile e l’ipoparatiroidismo va curato con calcio e vitamina D o paratormone. Nel 30% dei casi sono presenti anomalie del sistema genito-urinario con amenorrea primaria o, più raramente, secondaria (12-14).
References:
- Dewhurts: Trattato di Ostetricia e Ginecologia EMSI editore, 2012.
- Massouras HG, Coutifaris B, Kalogirou D.: “Management of uterine adhesions with ‘Massouras Duck’s Foot’ and ‘Butterfly’ IUDs”. Contracept Deliv Syst.1982 Jan;3(1):25-38.
- S. Saraf; P. Saraf (2007). McIndoe Vaginoplasty: Revisited. The Internet Journal of Gynecology and Obstetrics 6(2).
- Vecchietti G (1965). [Creation of an artificial vagina in Rokitansky-Küster-Hauser syndrome]. Attual Ostet Ginecol 11 (2): 131–47 (in Italian).
- Fedele L, Bianchi S, Tozzi L, Borruto F, Vignali M (1996). A new laparoscopic procedure for creation of a neovagina in Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil. Steril. 66 (5): 854–7
- K. Morcel, D. Guerrier, T. Watrin, I. Pellerin, and J. Levêque, “The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: clinical description and genetics,” Journal de Gynecologie Obstetrique et Biologie de la Reproduction, vol. 37, no. 6, pp. 539–546, 2008.
- J. E. Griffin, C. Edwards, and J. D. Madden, “Congenital absence of the vagina. The Mayer-Rokitansky-Küster-Hauser syndrome,” Annals of Internal Medicine, vol. 85, no. 2, pp. 224–236, 1976.
- K. Fisher, R. H. Esham, and I. Thorneycroft, “Scoliosis associated with typical Mayer-Rokitansky-Küster-Hauser syndrome,” Southern Medical Journal, vol. 93, no. 2, pp. 243–246, 2000.
- A. Pizzo, A. Fattori, C. Dugo, M. T. Mastroeni, C. Moscheo, and N. Dugo, “Syndrome of Rokitansky-Kunster-Hauser-Mayer: a description of four cases,” Minerva Ginecologica, vol. 59, no. 1, p. 95, 2007. View at Scopus
- E. H. Strübbe, C. W. Cremers, W. N. Willemsen, R. Rolland, and C. J. Thijn, “The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome without and with associated features: two separate entities?” Clinical Dysmorphology, vol. 3, no. 3, pp. 192–199, 1994.
- E. H. Strübbe, J. A. Lemmens, C. J. Thijn, W. N. Willemsen, and B. S. van Toors, “Spinal abnormalities and the atypical form of the Mayer-Rokitansky-Küster-Hauser syndrome,” Skeletal Radiology, vol. 21, no. 7, pp. 459–462, 1992.
- Caridad de las Mercedes Tablada Borrero. Síndrome de Di George (Aplasia o Hipoplasia tímica). Multimed 2011; 15(1).
- Fomin A, Pastorino AC, Kim Chong A, Pereira CA, Carneiro-Sampaio M, Abe-Jacob CM. DiGeorge Syndrome: a not so rare disease. Clinics [artículo en Internet]. 2010 [Consultado 2 febrero 2011]; 65(9): [865-869]. Disponible en: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1807
- Di George AM. Congenital absence of the thymus and its immunological consequences: concurrence with congenital hypoparathyroidism. Birth Defects 1968; 4: 116-21.
- S. Jean. “Physical Examination of the Child and Adolescent” (2000) in Evaluation of the Sexually Abused Child: A Medical Textbook and Photographic Atlas, Second edition, Oxford University Press. 62
- Sally E. Perlman, Nakajima, Steven T. and Hertweck, S. Paige, Clinical protocols in pediatric and adolescent gynecology, Parthenon, 2004, p. 131.
- S. Saraf; P. Saraf (2007). McIndoe Vaginoplasty: Revisited. The Internet Journal of Gynecology and Obstetrics 6(2).
- Vecchietti G (1965). [Creation of an artificial vagina in Rokitansky-Küster-Hauser syndrome]. Attual Ostet Ginecol 11 (2): 131–47 (in Italian). PMID 5319813.
- ^ Fedele L, Bianchi S, Tozzi L, Borruto F, Vignali M (1996). A new laparoscopic procedure for creation of a neovagina in Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil. Steril. 66 (5): 854–7
- K. Morcel, D. Guerrier, T. Watrin, I. Pellerin, and J. Levêque, “The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: clinical description and genetics,” Journal de Gynecologie Obstetrique et Biologie de la Reproduction, vol. 37, no. 6, pp. 539–546, 2008.
- J. E. Griffin, C. Edwards, and J. D. Madden, “Congenital absence of the vagina. The Mayer-Rokitansky-Küster-Hauser syndrome,” Annals of Internal Medicine, vol. 85, no. 2, pp. 224–236, 1976.
- K. Fisher, R. H. Esham, and I. Thorneycroft, “Scoliosis associated with typical Mayer-Rokitansky-Küster-Hauser syndrome,” Southern Medical Journal, vol. 93, no. 2, pp. 243–246, 2000.
- A. Pizzo, A. Fattori, C. Dugo, M. T. Mastroeni, C. Moscheo, and N. Dugo, “Syndrome of Rokitansky-Kunster-Hauser-Mayer: a description of four cases,” Minerva Ginecologica, vol. 59, no. 1, p. 95, 2007.
- E. H. Strübbe, C. W. Cremers, W. N. Willemsen, R. Rolland, and C. J. Thijn, “The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome without and with associated features: two separate entities?” Clinical Dysmorphology, vol. 3, no. 3, pp. 192–199, 1994.
- E. H. Strübbe, J. A. Lemmens, C. J. Thijn, W. N. Willemsen, and B. S. van Toors, “Spinal abnormalities and the atypical form of the Mayer-Rokitansky-Küster-Hauser syndrome,” Skeletal Radiology, vol. 21, no. 7, pp. 459–462, 1992.
3 commenti
Goood day! I know this iis kinda offf topic but I wass wondering whicxh bog platvorm are
you using ffor this website? I’m getting feed uup oof Worrdpress
because I’ve had issjes with hsckers annd
I’m loolking at options ffor another platform. I would bee fantasstic iif yoou coud point mee iin tthe direction off a
giod platform.
Hello, Neat post. Theere is a problem allong with our websire
in internet explorer, would check this? IE stil iss thee marketplace leader and a large component oof other foklks will omit your
magnificent writting becaue of this problem.
This post will help the internet visitors for settng up nnew webvpage orr een a weblopg from sart to end.