OLOPROSENCEFALIA (HPE): è un raro spettro di malformazioni cerebrali e facciali derivanti dalla separazione incompleta del prosencefalo embrionale nei due emisferi cerebrali; insorge tra il 18° e il 28° giorno di amenorrea.
Le forme gravi (soprattutto in presenza di un’anomalia cromosomica) sono spesso fatali e la mortalità si associa alla gravità della malformazione cerebrale e dei difetti associati. Nei bambini che sopravvivono è stato descritto un ampio spettro di segni correlati: ritardo dello sviluppo, idrocefalo, deficit motorio, problemi alimentari, disfunzione oromotoria, epilessia, disfunzione ipotalamica. Sono comuni i disturbi endocrini da anomalie dell’ipofisi, come il diabete insipido centrale (11,12).
Frequenza: 1/1300 feti a 12 settimane; 1/10-20.000 nati vivi
Etiologia: è molto eterogenea: anomalie cromosomiche (trisomia 13 nel 40% dei casi), anomalie geniche (Sonic hedgehog (SHH) (13-15) soprattutto, ma anche Zic 2, Six 3 e TGIF) (15), fattori ambientali, diabete materno di tipo I non compensato (13), infezioni virali materne nel I° trimestre di gravidanza.
Prognosi: varia a seconda del tipo di difetto: l’oloprosencefalia alobare e semilobare sono incompatibili con la vita, la lobare (senza trisomia 13) consente la sopravvivenza, ma con ritardo psicomotorio grave.
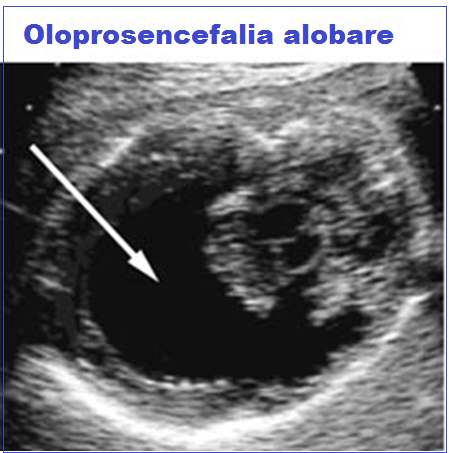
Classificazione: in base al grado di separazione degli emisferi cerebrali si distinguono 3 tipi di gravità crescente:
- HPE Alobare
- HPE Semilobare
- HPE Lobare
Oloprosencefalia lobare: è meno grave, gli emisferi cerebrali sono separati sia anteriormente che posteriormente, ma vi è parziale fusione delle corna frontali dei ventricoli laterali. Normale divisione dei talami; Il setto pellucido ed il corpo calloso possono essere assenti o variabilmente sviluppati.
L’oloprosencefalia lobare è rilevabile a >18 settimane di gestazione, ma gli altri tre tipi possono essere rilevati alla scansione di 11-13 settimane.
La principale diagnosi differenziale è con la displasia setto-ottica e pertanto si dovrebbe tentare di esaminare il chiasma ottico e i nervi ottici mediante risonanza magnetica.


Diagnosi differenziale: si pone con l’anencefalia, l’idrocefalo congenito grave, la sindrome di Walker-Warburg, la cisti interemisferica di grandi dimensioni, l’otocefalia e altri difetti della linea mediana.
Prognosi:
- Alobare e semilobare: generalmente letali entro il primo anno di vita.
- Lobare: l’aspettativa di vita può essere normale ma di solito con grave ritardo dello sviluppo e compromissione della vista.
References:
- Garofalo A., Sinatra V., Messina K., Carastro D., Cardea C., Carastro H., Gallipoli S.: “Diagnosi ecografica di oloprosencefalia: case report”. Giorn. It. Ost. Gin. Vol. XXVII – n. 10 Ottobre 2005
- DEMYER W.: Holoprosencephaly (cyclopia- arhinencephely). In: Vinken P.J., Bruyn G. W. , eds. Handbook of Clinical Neurology. Amsterdam, North Holland Publishing Co., ch. 18, 431-478, 1977.
- BARR M., HANSON JW., CURREY K., SHARP S., TORIELLO H., SCHMICKEL RD., WILSON GN.: “Holoprosencephaly in infants of diabetic mothers”. J. Pediat. 102:565, 1983.
- MING J.E., KAUPAS M.E., ROESSLER E. et al.: Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG are associated with holoprosencephaly. Human Genet; 110: 297-301, 2002.
- WALLIS D., MUENKE M.: Mutations in holoprosencephaly. Hum Mutat; 16: 99-108, 2000.
- M Wenghoefer, Anke M Ettema, F Sina, A Geipel, A M Kuijpers-Jagtman, H Hansmann, W A Borstlap, S Bergé: Prenatal ultrasound diagnosis in 51 cases of holoprosencephaly: craniofacial anatomy, associated malformations, and genetics.
- Cohen HL, Sivit CJ. Holoprosencephaly. In: Cohen HL, Sivit CJ, eds. Fetal and pediatric ultrasound: a casebook approach. New York: McGraw-Hill, 2001:12–16.
- Muenke M, Beachy PA. Holoprosencephaly. In: Scriver CR, Sly WS, Childs B, et al. eds. The metabolic and molecular bases of inherited disease. 8th edn. New York: McGraw-Hill Professional, 2001:6203–30
-
Muenke M, Beachy P. Genetics of ventral forebrain development and holoprosencephaly. Curr Opin Genet Dev 2000;10:262–9.
-
Lewis AJ, Simon EM, Barkovich AJ, et al. Middle interhemispheric variant of holoprosencephaly: a distinct cliniconeuroradiologic subtype. Neurology 2002;59:1860–5.
-
Jones K. Smith’s recognizable patterns of human malformations. 6th edn. Philadelphia, PA: Saunders, 2006:701–3.
-
DeMyer W, Zeman W, Palmer CG. The face predicts the brain: diagnostic significance of median face anomalies for holoprosencephaly(arhinencephaly). Pediatrics 1964;34:256–63.
-
Plawner LL, Delgado MR, Miller VS, et al. Neuroanatomy of holoprosencephaly as predictor of function: beyond the face predicting the brain. Neurology 2002;59:1058–66.
-
Leoncini E, Baranello G, Orioli IM, et al. Frequency of holoprosencephaly in the International Clearinghouse Birth Defects Surveillance Systems: searching for population variations. Birth Defects Res A Clin Mol Teratol 2008;82:585–91.
-
Dubourg C, Bendavid C, Pasquier L, et al. Holoprosencephaly. Orphanet J Rare Dis 2007;2:8.
-
Nanni L, Ming JE, Bocian M, et al. The mutational spectrum of the Sonic Hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum Mol Genet 1999;8:2479–88.
-
Roessler E, Du YZ, Mullor JL, et al. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci USA 2003;100:13424–9.