
Agenesia del corpo calloso
L’agenesia del corpo calloso colpisce lo 0.3-0.7% della popolazione generale. Essa consiste nella mancata formazione del corpo calloso, un ponte di tessuto nervoso, che mette in comunicazione i due emisferi. Può essere completa, o riguardare solo una parte del corpo calloso (agenesia parziale), solitamente la parte posteriore, che si forma più tardivamente.
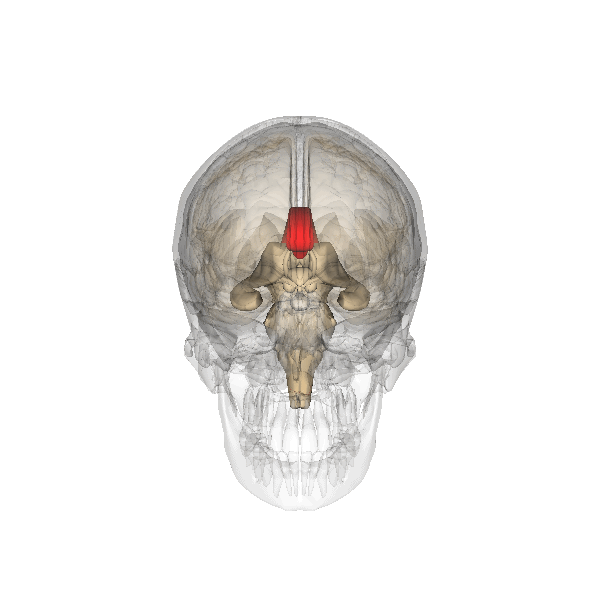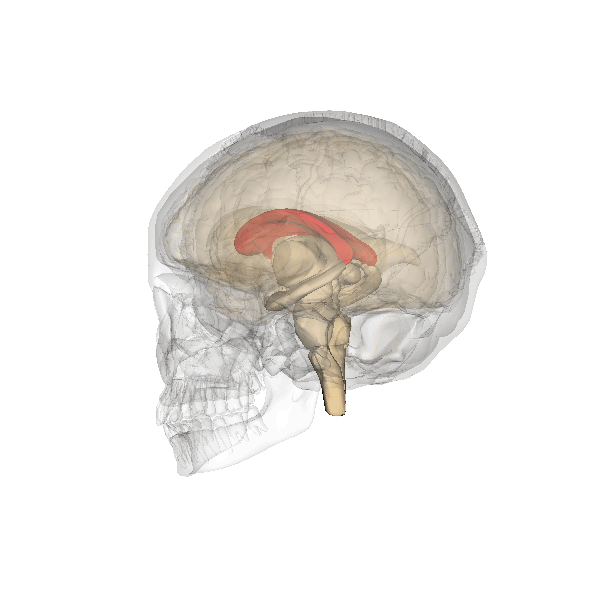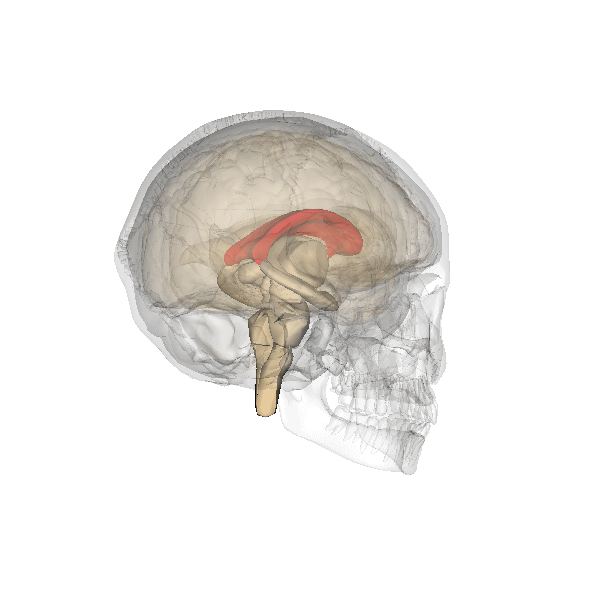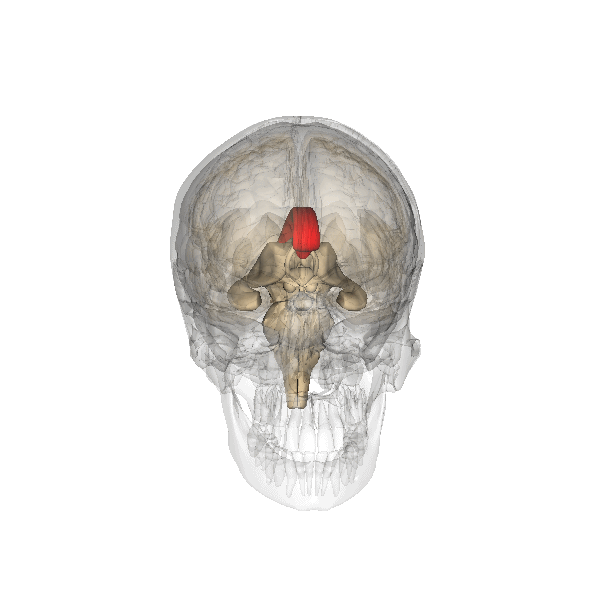
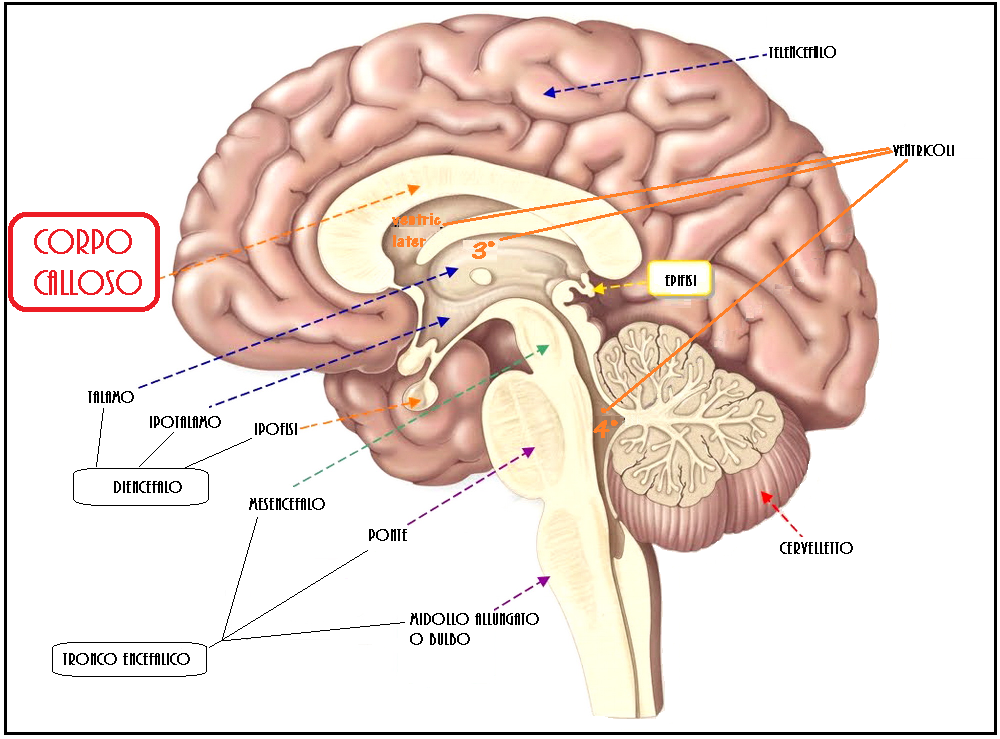
Corpo calloso
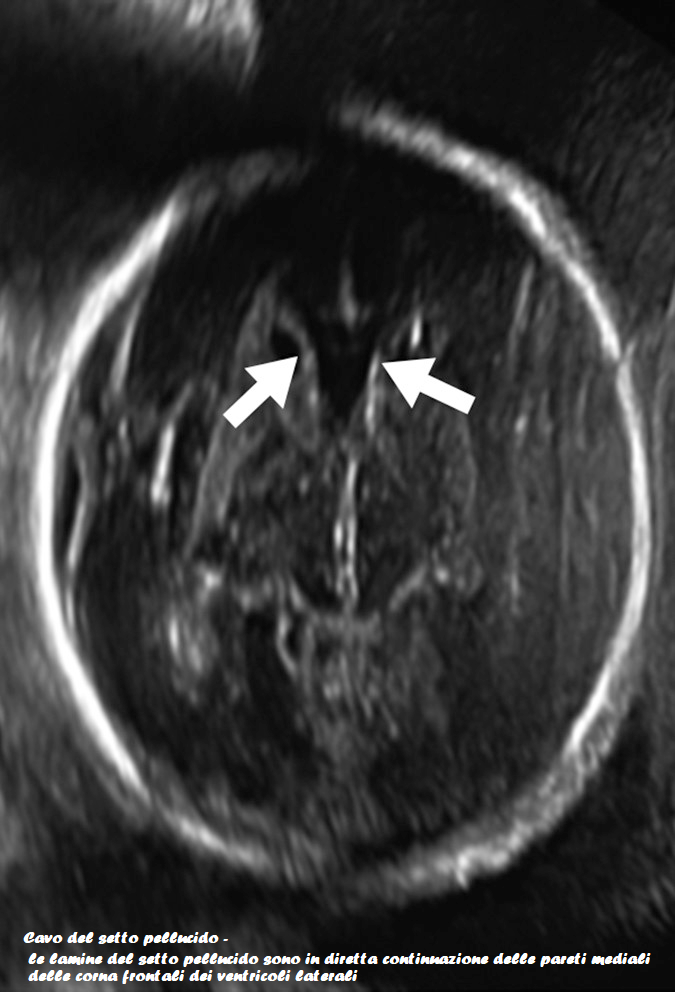
Il corpo calloso è un ponte di tessuto nervoso che mette in comunicazione i due emisferi cerebrali. Esso si sviluppa tra la 5a e la 18a settimana e pertanto la diagnosi di agenesia del corpo calloso non dovrebbe essere fatta prima della 19a w. In occasione dell’ecografia morfologica il corpo calloso si è già sviluppato, e la mancata visualizzazione del cavo del setto pellucido richiede ulteriore valutazione ecografica e con RMN.
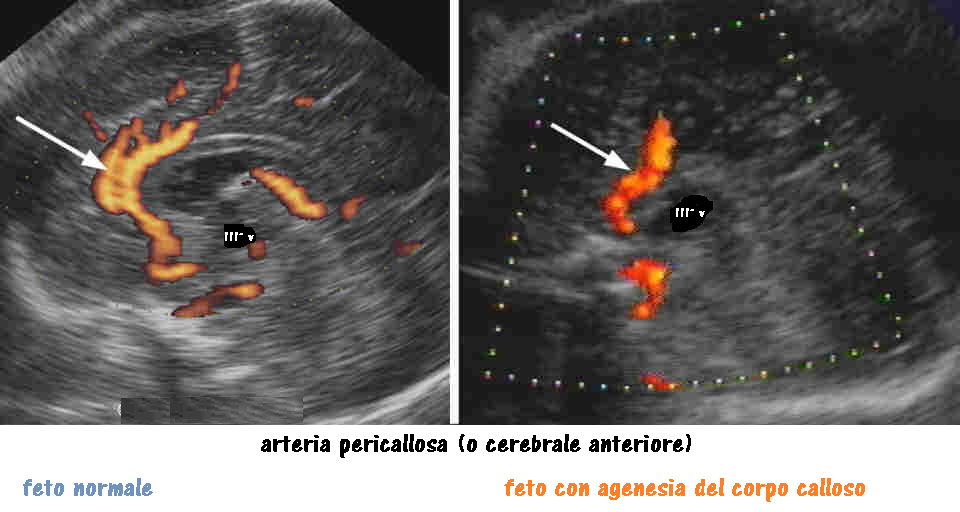
Ecograficamente l’agenesia del corpo calloso viene diagnosticata per l’assenza del cavo del setto pellucido, ventricolomegalia con ventricoli laterali a forma di goccia (“colpocefalia”) ed aumento della separazione dei corni anteriori dei ventricoli laterali. Il terzo ventricolo può apparire più in alto, essendo normalmente delimitato dal corpo calloso al di sopra. La scissura interemisferica può apparire prominente. In sezione sagittale, l’uso del color Doppler può aiutare a confermare la diagnosi, attraverso la visualizzazione di un decorso anomalo dell’arteria pericallosa, essendo assente il giro del cingolo (i giri corticali hanno un decorso anomalo ed appaiono radiati rispetto al terzo ventricolo).
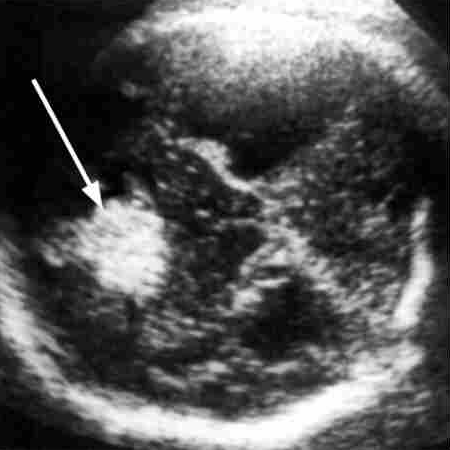
Lipoma intracranico
Anomalie associate sono presenti fino al 50% dei casi e comprendono: lipoma cerebrale (viene visualizzato come una massa iperecogena a livello della linea mediana), Acrocallosal syndrome, cisti interemisferica, malformazione di Dandy-Walker, eterotopia e anomalie della girazione, microcefalia e malformazioni di altri organi ed apparati (cardiaco, genito-urinario, ernia diaframmatica…).
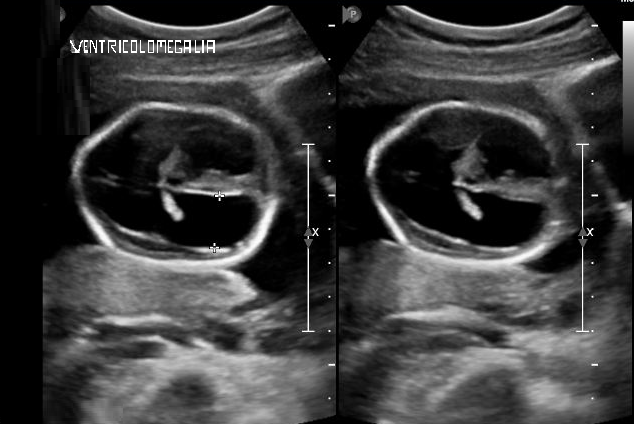
La ventricolomegalia – il diametro dei ventricoli laterali, a 20 settimane e misurato a livello dei corni occipitali, presenta un diametro <10 mm ed aumenta con l’epoca gestazionale. Oltre che all’agenesia completa del corpo calloso, la ventricolomegalia può essere associata a:
– Malformazioni del SNC: Dandy-Walker syndrome, Oloprosencefalia (S. di Andermann), Spina bifida.
– Fenomeni distruttivi di origine ischemica, emorragica o infettiva;
– Processi espansivi, tumorali e non;
– Sindromi polimalformative;
– Malattie genetiche;
– Cromosomopatie.
La diagnosi differenziale è con altre cause di ventricolomegalia e malformazioni che mostrano assenza del cavo del setto pellucido, quali l’oloprosencefalia lobare e la displasia setto-ottica.
La gestione di questa condizione prevede:
- ecografia di secondo livello, al fine di escludere altre malformazioni
- consulenza genetica, per discutere la possibile associazione con anomalie cromosomiche (10-20% dei casi, soprattutto trisomia 18, trisomia 13 e triplodia), sindromi genetiche ed esposizione a teratogeni (alcool, cocaina, acido valproico)
- amniocentesi
- risonanza magnetica fetale, per confermare la diagnosi ed evidenziare la presenza di malformazioni associate a carico del sistema nervoso centrale, in particolare per studiare la girazione corticale, un dato difficile da valutare ecograficamente
- ecografia 3D per ricostruire il piano sagittale del sistema nervoso centrale, se il feto è in posizione non favorevole
- consulenza con neurologo pediatra, per discutere la prognosi
La prognosi dipende dalla presenza di anomalie associate e dall’entità della ventricolomegalia. Lo sviluppo dei bambini con agenesia del corpo calloso isolata è normale nel 70% dei casi. Il rischio di ritardo psicomotorio è del 15% se dopo la nascita si conferma che l’agenesia del corpo calloso è realmente isolata, e non si associa ad altre anomalie (ad esempio sindromi genetiche) non possibili da diagnosticare in utero. L’agenesia del corpo calloso isolata ha un impatto limitato sulla capacità cognitiva, ed il quoziente intellettivo è solitamente nel range di normalità. I pazienti con questa patologia possono avere difetti del linguaggio espressivo, ridotte competenze sociali e bassa autostima.
Il rischio di ricorrenza in una successiva gravidanza è del 2-3%, ma può essere più alto se l’agenesia del corpo calloso rientra in una specifica sindrome genetica.