Ultimo aggiornamento 2022-03-03 22:45:36
La sindrome di Morris è una malattia rara che che si instaura durante lo sviluppo degli organi e degli apparati riproduttivo e genitale fetali. Consiste essenzialmente in una insensibilità dei testicoli all’azione degli androgeni con il risultato di sviluppare una persona fenotipicamente, e legalmente, donna con cariotipo maschile (45,XY).  Perciò è chiamata anche sindrome da femminilizzazione testicolare e anche sindrome da insensibilità agli androgeni (AIS, Androgen Insensitivity Syndrome) (1).
Perciò è chiamata anche sindrome da femminilizzazione testicolare e anche sindrome da insensibilità agli androgeni (AIS, Androgen Insensitivity Syndrome) (1).
Etiologia: L’insensibilità dei tessuti agli androgeni è causata da un allele recessivo (indicato con tfm) che si trova nel cromosoma X (X-linked). Altre mutazioni recettoriali sono state progressivamente scoperte ed indicate come la causa dell’insensibilità dell’azione androgenica (3-7)
E’ pertanto la madre che trasmette tale condizione. La prole geneticamente maschile sarà per il 50% affetta, mentre le figlie geneticamente XX saranno per il 50% portatrici. L’insensibilità dei tessuti bersaglio fetali all’azione degli ormoni androgeni ha come conseguenza lo sviluppo di gonadi maschili ritenute in addome, sviluppo caratteri sessuali femminili secondari (mammelle, no utero e ovaie!), ipoplasia vaginale (2,3).
Il processo della differenziazione sessuale è un sistema complesso di sviluppo regolato da fattori genetici e ormonali, che si svolge nelle prime fasi della gravidanza e usualmente si completa entro i primi 2-3 mesi di gravidanza. Al momento del concepimento si stabilisce infatti il sesso cromosomico del nuovo individuo: infatti se l’ovocita (che ha solo cromosomi sessuali X) viene fecondato da uno spermatozoo con X ne risulterà un embrione 46,XX (cioè con corredo cromosomico femminile); se invece viene fecondato da uno spermatozoo Y ne risulterà un embrione 46,XY (cioè con corredo cromosomico maschile). Indipendentemente dall’assetto cromosomico, nelle prime fasi di sviluppo embrionario, fino all8a w sia le gonadi primitive sia l’aspetto dell’apparato genitale sono uguali. Con il procedere dello sviluppo dell’embrione, la gonade primitiva subisce una serie di trasformazioni che la rendono differente tra i due sessi, sia per quanto riguarda gli aspetti morfologici sia per la funzione endocrina. In particolare, il gene SRY (Sex determining Region of Y chromosome) presente sul braccio corto del cromosoma Y determina, anche con la cooperazione con altri geni, la differenziazione della gonade primitiva in testicolo nei feti con cariotipo 46,XY.
Il testicolo fetale produce degli ormoni che determinano la modificazione dell’apparato riproduttivo (cioè degli organi genitali interni ed esterni) in senso maschile.Come accennato, fino alla 8° settimana di gestazione l’apparato riproduttivo è del tutto uguale in ambedue i sessi e di “aspetto femminile”. Nei feti in cui si è formato un testicolo si ha una trasformazione degli organi genitali interni ed esterni in senso maschile per l’azione principalmente di due ormoni: l’ormone antimulleriano, prodotto dalle cellule di Sertoli e il testosterone, prodotto dalle cellule di Leydig, e del suo metabolita DHT, più attivo, prodotto direttamente nei tessuti bersaglio per l’azione dell’enzima 5-alfa-reduttasi.
Se l’ormone antimulleriano (AMH), il testosterone o il diidrotestosterone (DHT) non vengono prodotti o non riescono a funzionare, totalmente o parzialmente, il feto non potrà sviluppare le strutture riproduttive maschili e presenterà alla nascita un aspetto femminile o con vari gradi di ambiguità dei genitali. Il testosterone è coinvolto principalmente nello sviluppo e differenziazione delle strutture derivate dai dotti di Wolff, mentre il 5-alfa-diidrotestosterone (DHT), è il ligando attivo sul seno urogenitale, il tubercolo e le strutture derivate (8). Il gene del recettore degli androgeni si trova sul cromosoma X e precisamente sui loci Xq11–12 e codifica per una proteina con una massa molecolare di circa 110 kDa. Sono state identificate finora 6 mutazioni del gene del recettore per gli androgeni.
La sindrome da insensibilità agli androgeni (Androgen Insensitivity Syndrome, AIS), nota in precedenza come femminilizzazione testicolare, provoca un’interruzione dello sviluppo dell’apparato riproduttivo nel feto maschio. Nel suo corpo si sviluppano i testicoli embrionali che iniziano a produrre androgeni. Ma tali ormoni maschili non possono completare uno sviluppo dei caratteri maschili a causa di una rara insensibilità dei tessuti fetali agli androgeni. Per questo motivo, le caratteristiche genitali esterne si sviluppano seguendo le linee femminili (come sistema di emergenza), dopo che lo sviluppo degli organi femminili interni è già stato interrotto dall’AMH prodotto nel feto dai testicoli.
L’insensibilità dei tessuti agli androgeni è causata da un gene che si trova nel cromosoma sessuale X e l’AIS è una condizione recessiva legata al cromosoma X da parte della madre (in circa un terzo dei casi, questa condizione è il risultato di una mutazione spontanea). Una madre portatrice di un gene difettoso ha una possibilità su due che suo figlio XY sia affetto da AIS e una possibilità su due che una figlia XX sia lei stessa portatrice di un gene difettoso. Le donne XX possono sottoporsi a test specifici per verificare se sono portatrici.
L’AIS è uno fra i numerosi “Disordini della Differenziazione Sessuale” (DSD) Può essere considerato una forma intersessuale in quanto c’è un disaccordo tra il sesso genetico e la formazione dei genitali esterni.
Questa condizione tuttavia è differente dalla transessualità. Le affette da questa sindrome sono dal punto di vista anatomico, psicologico, legale e sociale delle donne.
L’AIS è una condizione poco frequente e perciò è inclusa nella lista delle malattie rare del Ministero della Sanità. La condizione di “Malattia Rara” consente l’esenzione dal ticket con il codice RNG010 (RNG010); nella lista del Ministero l’AIS è presente con la vecchia denominazione “Pseudoermafroditismi”
Classificazione: l’insensibilità agli androgeni può essere di vari gradi, da 1 (leggera) a 7 (totale o completa). Se l’insensibilità agli androgeni è completa (grado 7) si parla di CAIS. Se invece l’insensibilità è parziale (gradi da 1 a 6) si parla di PAIS. Nel PAIS sono quindi comprese situazioni cliniche e fenotipiche diverse a seconda : della gravità del grado:
Grado 1-2: i genitali esterni appaiono come maschili a tutti gli effetti, ma già al livello 2, i genitali hanno le caratteristiche di quelli di un maschio con ipospadia, ovvero apertura uretrale collocata nella superficie inferiore del pene, e in alcuni casi questa apertura – canale può arrivare fino al glande.
Grado 3: si hanno forme più mascolinizzate del grado 4, le grandi labbra sono comunque fuse e lo stoma uretrale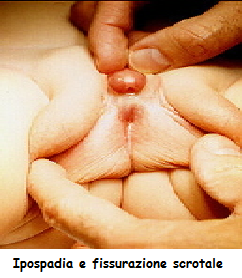 si trova alla base del clitoride – pene.
si trova alla base del clitoride – pene.
Gradi 4-5: troviamo l’evidente presenza di genitali femminili, con il clitoride che si presenta di grandi dimensioni Nel grado 5, inoltre, si può avere una parziale fusione delle grandi labbra, che formano una membrana a copertura dell’apertura della vagina. Nel grado 4 questa membrana di tessuto può arrivare a coprire anche l’uscita dell’uretra. La cavità che ne deriva si chiama seno urogenitale.
Diagnosi:
- l’amenorrea è quasi sempre il motivo per cui i genitori della paziente richiedono una consulenza ginecologica e pediatrica.
- Fenotipo: nella classica sindrome completa (CAIS), la paziente presenta un fenotipo femminile, gradevole (sindrome delle belle donne), altezza media o superiore alla norma, cute marmorea, mammelle ben sviluppate, peli pubici presenti e di tipo femminile, peli ascellari assenti.
- Vagina: L’esame fisico delle pazienti rivela tipicamente una vagina cieca. Generalmente nel caso di AIS, il terzo superiore della vagina può risultare mancante, ma in alcuni casi, la vagina potrebbe non superare 1 o 2 cm di lunghezza. I medici non devono tralasciare di esaminare la vagina nelle pazienti con AIS durante la pubertà, poiché alcune adolescenti potrebbero scoprire il problema da sole, nasconderla e vivere con questo segreto nella paura e nell’isolamento per anni.
- ernia inguinale bilaterale.
- Cariotipo: determina il sesso gonadico e induce a ricercare l’insensibilità agli androgeni.
- USG: Lo studio ecografico dell’addome conferma la diagnosi rilevando le gonadi ritenute e l’assenza di utero ed ovaie.

Management: Molti pediatri raccomandano l’analisi cromosomica per ogni ragazza che evidenzi all’esame clinico un’ernia inguinale. Se viene individuato un cromosoma Y, è consigliabile procedere alla  gonadectomia immediatamente a causa del rischio di degenerazione neoplastica (gonadoblastoma, teratoblastoma e, molto raramente cistoadenofibroma papillifero (9), cisti gonadali (10)). Questa opinione trova però dissenzienti numerosi AA. che consigliano di operare una vigile attesa almeno fino al compimento della pubertà (18-20 anni) confrontando il basso il rischio di degenerazione neoplastica e gli indubbi vantaggi di una buona impregnazione ormonale. Nelle donne CAIS, infatti, i testicoli producono oltre agli androgeni, ai quali l’organismo Morris è insensibile, anche gli estrogeni che sviluppano la loro azione sull’organismo favorendo la crescita e la maturazione dei caratteri sessuali secondari femminili. Dopo la pubertà molt AA. consigliano comunque di evitare la gonadectomia considerando che il rischio di degenerazione neoplastica è molto basso nelle pazienti CAIS. Invece, in caso di PAIS, dopo la pubertà è assolutamente necessario procedere con la gonadectomia a causa dell’elevato rischio neoplastico e della parziale sensibilità agli androgeni.
gonadectomia immediatamente a causa del rischio di degenerazione neoplastica (gonadoblastoma, teratoblastoma e, molto raramente cistoadenofibroma papillifero (9), cisti gonadali (10)). Questa opinione trova però dissenzienti numerosi AA. che consigliano di operare una vigile attesa almeno fino al compimento della pubertà (18-20 anni) confrontando il basso il rischio di degenerazione neoplastica e gli indubbi vantaggi di una buona impregnazione ormonale. Nelle donne CAIS, infatti, i testicoli producono oltre agli androgeni, ai quali l’organismo Morris è insensibile, anche gli estrogeni che sviluppano la loro azione sull’organismo favorendo la crescita e la maturazione dei caratteri sessuali secondari femminili. Dopo la pubertà molt AA. consigliano comunque di evitare la gonadectomia considerando che il rischio di degenerazione neoplastica è molto basso nelle pazienti CAIS. Invece, in caso di PAIS, dopo la pubertà è assolutamente necessario procedere con la gonadectomia a causa dell’elevato rischio neoplastico e della parziale sensibilità agli androgeni.
Terapia sostitutiva ormonale: Quando le gonadi vengono rimosse dopo la pubertà, è necessario ricorrere a una terapia sostitutiva a base di ormoni femminili a lungo termine per prevenire i sintomi della menopausa, contro l’osteoporosi e per proteggere le pazienti dalle malattie cardio-vascolari. Quando l’asportazione delle gonadi viene eseguita nel periodo dell’infanzia o della fanciullezza, la terapia viene iniziata solitamente a 10 o 11 anni, per permettere l’inizio della pubertà.
Prevenzione osteoporosi: La ridotta densità ossea pare sia più frequente nelle donne con AIS rispetto alle donne XX. La causa non è chiara. L’assenza di una terapia ormonale sostitutiva è un fattore di rischio, anche se alcune pazienti adulte con AIS presentano una ridotta densità ossea nonostante la terapia ormonale. Probabilmente ciò è dovuto al fatto che, durante il periodo in cui dovrebbe avvenire la costruzione delle ossa sane, le ‘ragazze XY’ producono un tasso di estrogeni inferiore a quello delle ragazze XX con le ovaie. Nelle ragazze XX la produzione di estrogeni inizia all’incirca a 8 anni (vale a dire 1 o 2 anni prima dello sviluppo mammario), per questo motivo potrebbe essere consigliabile la somministrazione di piccole dosi supplementari di estrogeni nelle pazienti con AIS a partire da questa età (1). Tuttavia, l’insensibilità agli androgeni è essa stessa responsabile della bassa densità ossea nonostante l’assunzione di estrogeni.
Vaginoplastica: Sull’intervento di plastica vaginale c’è unanimità di consenso. Si può risolverla con apposite tecniche chirurgiche, illustrate in altro capitolo.
.

![]()
References:
- Greenspan – Endocrinologia generale e clinica – Piccin
-
Dell’Edera D1, Malvasi A, Vitullo E, Epifania AA, Tinelli A, Laterza F, Novelli A, Pacella E, Mazzone E, Novelli G. Androgen insensitivity syndrome (or Morris syndrome) and other associated pathologies. Eur Rev Med Pharmacol Sci. 2010 Nov;14(11):947-57.
-
Raicu F1, Giuliani R, Gatta V, Palka C, Franchi PG, Lelli-Chiesa P, Tumini S, Stuppia L. Novel mutation in the ligand-binding domain of the androgen receptor gene (l790p) associated with complete androgen insensitivity syndrome. Asian J Androl. 2008 Jul;10(4):687-91.
-
Jääskeläinen J1, Mongan NP, Harland S, Hughes IA. Five novel androgen receptor gene mutations associated with complete androgen insensitivity syndrome. Hum Mutat. 2006 Mar;27(3):291.
-
McPhaul MJ Androgen receptor mutations and androgen insensitivity. Mol Cell Endocrinol. 2002 Dec 30;198(1-2):61-7.
-
Narumi S, Amano N, Hachiya R, Ishii T, Hasegawa T. A novel mutation of androgen receptor gene in complete androgen insensitivity syndrome. Clin Pediatr Endocrinol. 2007;16(2):59-61. doi: 10.1297/cpe.16.59. Epub 2007 May 17.
-
Sultan C, Lumbroso S, Poujol N, Belon C, Boudon C, Lobaccaro JM.Mutations of androgen receptor gene in androgen insensitivity syndromes. J Steroid Biochem Mol Biol. 1993 Nov;46(5):519-30.
-
Brinkmann AO Molecular basis of androgen insensitivity. Mol Cell Endocrinol. 2001 Jun 20;179(1-2):105-9.
-
Ozdemir O, Sari ME, Akmut E, Selimova V, Unal T, Atalay CR. Complete androgen insensitivity syndrome with a large gonadal serous papillary cystadenofibroma. J Hum Reprod Sci. 2014 Apr;7(2):148-50. doi: 10.4103/0974-1208.138875.
- Yanai Y, Hiroi H, Osuga Y, Fujimoto A, Momoeda M, Yano T, Taketani Y. Androgen insensitivity syndrome with serous gonadal cyst. Fertil Steril. 2008 Nov;90(5):2018.e9-11.
7 commenti
Hello, yup this article is genuinely fastidious and I have learned lot of things from it on the topic of blogging.
thanks.
Wow, this article is fastidious, my younger sister is analyzing these kinds of things, thus I am going to tell her.
Hey There. I found your blog the usage of msn.
That is an extremely well written article.
I will be sure to bookmark it and return to learn more of your helpful info.
Thanks for the post. I’ll certainly return.
Thank you for sharing your info. I really appreciate
your efforts and I am waiting for your further write ups thank you once again.
Hi there, I enjoy reading all of your article.
I wanted to write a little comment to support you.
You are so cool! I don’t suppose I have read anything like that before.
So nice to discover somebody with genuine thoughts on this topic.
Really.. many thanks for starting this up. This web site is one thing that’s needed on the internet, someone with a little originality!
This design is steller! You certainly know how to
keep a reader amused. Between your wit and your videos,
I was almost moved to start my own blog (well, almost…HaHa!) Great
job. I really enjoyed what you had to say, and more than that, how you
presented it. Too cool!