L’ipogonadismo maschile è una condizione patologica in cui un maschio non produce adeguate quantità di testosterone o presenta alterazioni recettoriali allo stesso testosterone pur presente in circolo in quantità normali. Il testosterone è un ormone chiave dell’organismo maschile, responsabile per la crescita, il normale sviluppo fisico durante gli anni della pubertà e indispensabile anche per la spermatogenesi (1,2).
Le gonadi maschili sono sotto il controllo dell’asse ipotalamo-ipofisario mediante il Gn-RH  ipotalamico che, secreto in maniera pulsatile ogni 2 ore circa, agisce sulle cellule gonadotrope dell’ipofisi stimolandone la sintesi e secrezione di LH ed FSH. L’FSH agisce stimolando la spermatogenesi a livello delle cellule del Sertoli. L’LH agisce sulle cellule del Leydig promuovendo la secrezione di testosterone e inibina. Il testosterone, a sua volta, esercita un feedback negativo sia a livello ipofisario, sia a livello ipotalamico mentre l’inibina deprime in maniera selettiva la secrezione di FSH, senza influenzare quella dell’LH.
ipotalamico che, secreto in maniera pulsatile ogni 2 ore circa, agisce sulle cellule gonadotrope dell’ipofisi stimolandone la sintesi e secrezione di LH ed FSH. L’FSH agisce stimolando la spermatogenesi a livello delle cellule del Sertoli. L’LH agisce sulle cellule del Leydig promuovendo la secrezione di testosterone e inibina. Il testosterone, a sua volta, esercita un feedback negativo sia a livello ipofisario, sia a livello ipotalamico mentre l’inibina deprime in maniera selettiva la secrezione di FSH, senza influenzare quella dell’LH.
La riduzione o la completa assenza della produzione di androgeni si associa sempre ad una ridotta spermatogenesi perchè l’alta concentrazione di testosterone a livello dei tubuli seminiferi è fondamentale per una normale spermatogenesi sia all’inizio della pubertà sia nel corso della vita riproduttiva. Al contrario, la funzione endocrina testicolare può essere normale anche in assenza di spermatogenesi.
CLASSIFICAZIONE:
- IPOGONADISMO PRIMARIO O IPERGONADOTROPO: si riscontrano bassi livelli plasmatici di androgeni e alti livelli di gonadotropine per alterazione della steroidogenesi testicolare che a sua volta può essere congenita o secondaria ad infezioni, flogosi o traumi.
- IPOGONADISMO SECONDARIO O IPOGONADOTROPO: si evidenzia una diminuzione sia degli androgeni che delle gonadotropine per alterazioni dell’asse ipotalamo-ipofisario.
- IPOGONADISMO DA RESISTENZA PERIFERICA: il problema è a carico o della 5-α-reduttasi (enzima che converte il testosterone in DHT) o del recettore degli androgeni: insensibilità recettoriale agli androgeni rappresentato in forma incompleta nella sindrome di Reifenstein e nella sindrome di Morris (o femminilizzazione testicolare o AIS) in cui si può osservare una forma completa di insensibilità recettoriale agli androgeni.
- alterazione genetica per cui vengono prodotti 2 o più cromosomi X come si verifica nella sindrome di Klinefelter.
- Infezione da virus della parotite durante l’adolescenza o nell’età adulta
- Emocromatosi con conseguente lesioni testicolari
- Chemioterapia e radioterapia
- Sindrome di Kallmann
- Sindrome di Sheehan (emorragie, traumi, cancro ipofisario)
- TBC
- AIDS
- Antinfiammatori
- Normale processo di invecchiamento
Fattori di rischio:
- traumi testicolari
- criptorchidismo infantile
Sintomatologia: è molto variabile e dipende da diversi fattori: epoca di comparsa del deficit (durante il periodo fetale, prima della pubertà, dopo la pubertà); entità del deficit; eziopatogenesi (primaria, secondaria, da resistenza); tipo di funzione alterata (tubulare, interstiziale o entrambe).
In caso di ipogonadismo prenatale, il feto non produce adeguate quantità di testosterone (T) che normalmente è sintetizzato dal feto già a partire dalla sesta settimana di gestazione in quantità di 0,5 mg/die. In questo stadio il T promuove la crescita ossea e muscolare ed è responsabile della differenziazione sessuale. Il neonato presenterà alterazioni della differenziazione dei genitali esterni e/o interni (a seconda del periodo della gravidanza in cui si è avuto il deficit); se si ha un deficit totale, si svilupperà un individuo che è cromosomicamente maschio, ma fenotipicamente femmina. Se il deficit è minore, invece, il neonato presenterà un’ambiguità sessuale o genitali maschili iposviluppatim in rapporto all’entità del deficit di testosterone, e pubertà ritardata.
In caso di ipogonadismo prepuberale, il pz presenterà scarso sviluppo dei caratteri sessuali secondari e sterilità. Vi possono essere anche turbe comportamentali, che rientrano in quello che prende il nome di infantilismo psichico, con alterata maturazione e scarso desiderio in ambito sessuale. Il pz, poi, presenterà delle ossa particolarmente lunghe per via della mancata chiusura delle cartilagini ipofisarie (con apertura delle braccia >5 cm rispetto all’altezza), ritardo dello sviluppo della massa muscolare, mancato approfondimento della voce, scarsa peluria maschile, iposviluppo del pene e dei testicoli, ginecomastia.
In caso di ipogonadismo postpuberale, il pz presenterà diminuzione della libido, infertilità e disfunzione erettile, scarso sviluppo dei peli di tipo maschile, diminuzione della massa muscolare, facile affaticamento, difficoltà di concentrazione e vampate. In condizioni gravi, si può verificare anche una regressione dei caratteri sessuali secondari.
La presentazione del quadro clinico, inoltre, varia da caso a caso: nella forma secondaria, ad esempio, possono associarsi deficit di altre tropine ipofisarie a causa di un tumore, o di lesione traumatica, o di un problema congenito, che ovviamente condizionano la sintomatologia.
Diagnostica di laboratorio:
Ipogonadismo primario o ipergonadotropo: oltre alla presenza di oligo-azoospermia, basse concentrazioni di testosterone ed elevata ipergonadotropinemia in conseguenza dell’assenza del feedback negativo da parte del testosterone sull’ipofisi. L’esame del cariotipo, che ci si aspetta essere XY, è importante anche per valutare l’eventuale presenza di alterazioni cromosomiche o mutazioni geniche. In presenza di oligo-azoospermia, con il solo FSH elevato e LH e testosterone normali, è bene indirizzarsi verso il solo deficit tubulare. – Biopsia testicolare.
Ipogonadismo secondario o ipogonadotropo: Oltre alla presenza di oligo-azoospermia, si avranno bassi valori sia di gonadotropine che di testosterone; La presenza di ridotti livelli di una sola delle due gonadotropine (LH o FSH) con valori normali o elevati dell’altra possono suggerire un quadro di difetto di produzione e secrezione di una sola gonadotropina e andrà ulteriormente indagata da un punto vista dinamico, biopsia testicolare, determinazione della HPRL ed esame radiodiagnostico con RMN della regione sellare, per valutare l’eventuale presenza di tumori o alterazioni congenite a carico dell’ipofisi. Può essere effettuato anche il test di stimolo con GnRH, sia per valutare l’entità del deficit gonadotropinico, sia per distinguere le forme di ipogonadismo secondario ipofisario da quello ipotalamico (1-11).
Diagnosi differenziale della S. di Kallmann: con altre forme di ipogonadsmo ipogonadotropo di origine ipotalamica, tra cui quelle secondarie a disturbi psichiatrici (ad es. anoressia nervosa), a 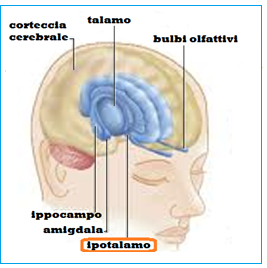 stress psico-fisico (ad es. atleti, calo ponderale, etc), o difetti isolati (non associati ad anosmia) della secrezione o azione del GnRH (13-17).
stress psico-fisico (ad es. atleti, calo ponderale, etc), o difetti isolati (non associati ad anosmia) della secrezione o azione del GnRH (13-17).
Variazioni della secrezione di testosterone in rapporto all’età: Il testosterone viene sintetizzato già dal feto (a partire dalla sesta settimana di gestazione) in quantità intorno a 0,5 mg/die. In questo stadio promuove la crescita ossea e muscolare ed è responsabile della differenziazione sessuale.
- Aumenta lentamente fino a circa 1 mg/die entro i dieci anni di età.
- Entro i dieci e vent’anni di età (adolescenza maschile) la sintesi aumenta rapidamente fino a raggiungere 5 ÷ 7 mg/die per rimanere a questo livello fino a circa trent’anni.
- Dopo i trent’anni, la sintesi diminuisce di circa 2% all’anno fino a raggiungere 3-4 mg/die all’età di ottant’anni.
Si notano differenze individuali di ±15% tra individui poco o molto virili: un maschio poco virile raggiunge a vent’anni una produzione testosteronica → pari a quella di cui un maschio molto virile dispone ancora a sessant’anni.
Le sieroconcentrazioni non sono “parallele” alla sintesi, perché oltre alla quantità di testosterone sintetizzato subentrano altrettanto complessi meccanismi di trasformazione e di smaltimento metabolico sulla concentrazione ematica.
Variazioni circadiane della concentrazione ematica del testosterone: Il testosterone è sintetizzato dalle cellule di Leydig nell’interstizio testicolare a partire dal colesterolo. La maggior parte si lega poi all’albumina e al SHGB (sex hormone-binding globulin) ematica.
La metabolizzazione è caratterizzata da due meccanismi:
- conversione periferica (negli organi bersaglio) in DHT ed estradiolo
- decomposizione nel fegato in diversi metaboliti; congiunzione e smaltimento renale come 17-keto-steroide.
Inoltre, a causa di “sfasamenti” di processi di sintesi e di conversione / smaltimento c’è una grande variazione circadiana: la testosteronemia raggiunge un minimo verso la 1:00 di notte. Poco dopo, la regolazione causa un notevole aumento della secrezione mentre la catabolizzazione diminuisce, il che fa rapidamente aumentare la testosteronemia fino alle 6:00 ÷ 12:00. nelle ore pomeridiane prevalgono i processi metabolici e la testosteronemia si abbassa lentamente fino alla 1:00 di notte.
Valori di riferimento
Non è stato ancora stabilito un limite inferiore “normale” del testosterone, si ritiene corretto riferirsi al range di variabilità illustrato nella seguente tabella:
| Testosterone totale | Testosterone libero | |
| > 12 nmol/l (346 ng/dl) | > 250 pmol/l (72 pg/ml) | valori ottimali |
| < 8 nmol/l (231 ng/dl) | < 180 pmol/l (52 pg/ml) | deficit |
Sarebbe utile eseguire il prelievo il mattino (tra le ore 7:00 e le ore 11:00) ed eventualmente in due prelievi separati per la variabilità ultradiana e circadiana.
Prolattina: Il dosaggio della PRL è utile per evidenziare ipogonadismo centrale da iperprolattinemia, mentre il dosaggio degli altri ormoni adeno-ipofisari è importante per escludere eventuali deficit ormonali multipli o ipersecrezione da adenomi ormono-secernenti.
Test di stimolo con Gn-RH:
Bassi livelli sierici di FSH, LH e testosterone: in tal caso un test di stimolo con Gn-RH esogeno alla dose di 100 µg ev in bolo permette di valutare la riserva ipofisaria delle gonadotropine. In generale i livelli di LH presentano un incremento di circa 2-5 volte mentre quelli di FSH di circa 2 volte. Questo test non è però utile nella diagnosi differenziale tra pubertà ritardata e ipogonadismo centrale in quanto ancora una volta i dati ormonali possono essere sovrapposti. Il suo impiego può essere giustificato per la valutazione di quadri di deficit singolo delle gonadotropine, per evidenziare il mancato aumento della gonadotropina deficitaria in presenza di un normale aumento dell’altra gonadotropina (18). D’altro canto può essere utile nel discriminare tra un ipogonadismo centrale di origine ipofisaria o ipotalamica benché, anche in casi di deficit ipotalamico di lunga durata, la risposta ipofisaria può essere ridotta per scarsa capacità delle cellule gonadotrope di rispondere allo stimolo in acuto. In questi casi è più utile la stimolazione con GnRH dopo boli ripetuti con microiniettore computerizzato.
Esami complementari:
- RMN dell’encefalo con particolare attenzione alla regione ipotalamo-ipofisaria è di
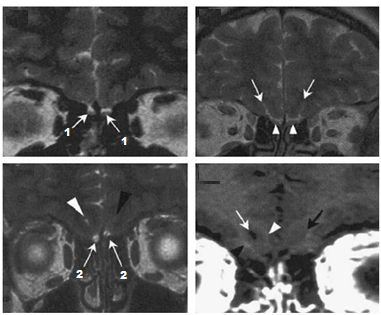 fondamentale importanza nello studio di queste forme centrali di ipogonadismo e permette di rilevare ipoplasia del bulbo olfattivo o anomalie alla base di alcune forme acquisite. D’altro canto, nell’ambito delle forme congenite, la RMN delle strutture olfattorie (bulbi, tratti e solchi) permette di confermare il sospetto diagnostico di sindrome di Kallmann.
fondamentale importanza nello studio di queste forme centrali di ipogonadismo e permette di rilevare ipoplasia del bulbo olfattivo o anomalie alla base di alcune forme acquisite. D’altro canto, nell’ambito delle forme congenite, la RMN delle strutture olfattorie (bulbi, tratti e solchi) permette di confermare il sospetto diagnostico di sindrome di Kallmann. - test olfattometrici tra cui il University of Philadelphia Inventory Sensory Test (UPSIT, in cui viene testata la sensibilità a 40 sostanze diverse e che risulta positivo per il riconoscimento di un numero <35/40.
- USG: per valutare il volume gonadico nei soggetti prepuberi e per la valutazione delle logge renali nei casi di sindrome di Kallmann. Infine, è utile indirizzare i pazienti presso centri specializzati per l’esecuzione di specifiche analisi genetiche, al fine di identificare eventuali difetti a carico dei geni riportati in letteratura e fornire un adeguato counselling genetico.
Terapia:
La terapia dell’ipogonadismo centrale isolato è volta al ripristino di valori di ormoni sessuali il più possibile prossimi ai livelli fisiologici, nell’induzione ormonale della pubertà e nel ripristino della fertilità. Non è disponibile una terapia per l’anosmia. Se l’ipogonadismo non viene adeguatamente trattato, l’individuo sviluppa un habitus eunucoide (12). Nelle forme in cui l’pogonadismo centrale è secondario ad altra condizione patologica e/o causale sarà necessario, qualora possibile, rimuovere tale causa.
- HMG: A differenza delle forme di ipogonadismo primitivo, nel caso di ipogonadismo centrale è possibile eseguire terapie con gonadotropine (HMG 150 UI ogni 3 giorni) al fine di indurre la spermatogenesi e la produzione degli androgeni (19).
- Gn-RH somministrato in maniera pulsatile, per via sottocutanea per mezzo di un microinfusore computerizzato portatile per infusione pulsatile ev/sc (Gonadorelina, Lutrelef® flac 10.8 mg/10 ml = 8.0 mg di farmaco/flac). Le dosi variano da 1 a 30 µg per bolo con una frequenza di somministrazione fra i 60 ed i 180 minuti.
- Clomifene citrato: in alternativa al testosterone per il trattamento dell’ipogonadismo in giovani pazienti. La scelta del clomifene potrebbe rivelarsi importante per risparmiare i testicoli dall’atrofia indotta dal testosterone esogeno (20).
- FIVET/ICSI
- Testosterone: nei pazienti che non desiderano fertilità, ed in cui non si sono manifestate segni di ripresa della funzionalità ipotalamo-ipofisaria dopo terapia con Gn-Rh o gonadotropine, la terapia dell’ipogonadismo è quella sostitutiva con testosterone i.m. (Testogen rapid 200 mg/ml fl 1 mg) o per via transdermica sia in cerotti (patch) che in gel poichè la cute assorbe in modo adeguato i composti steroidei. Successivamente alla modalità di applicazione a livello scrotale, non esente da inconvenienti, quali necessità di rasatura locale e difficoltosa aderenza in caso di testicoli di volume ridotto (evenienza non rara in pazienti ipogonadici), dal 1995 si è reso disponibile sul mercato il cerotto ad applicazione non scrotale (da posizionare a livello di braccia, regione lombare, addome). Tale modalità di somministrazione ha spesso presentato però, come effetto collaterale, l’irritazione cutanea nel sito di applicazione (con un’incidenza fino al 60%), la quale è stata sovente causa di interruzione del trattamento.A partire dal 2000, è stato messo in commercio un gel idroalcolico a base di T (25 o 50 mg di T per 2.5 o 5 g di gel) ad applicazione quotidiana su cute glabra, fino al completo assorbimento, preferibilmente sempre alla stessa ora del mattino. L’assorbimento è pari al 10-15% del T contenuto nella dose applicata. Tale modalità di somministrazione mantiene i livelli di T sierico nell’intervallo di normalità già dopo un’ora dall’applicazione con raggiungimento di un livello costante dopo 48-72 ore dall’inizio della terapia. Analogamente, l’interruzione della terapia riduce le concentrazioni di T ai livelli di pre-trattamento dopo 2 -3 giorni. La maneggevolezza della somministrazione di T a breve durata di azione come questa di cui abbiamo finora discusso, con pronto ripristino dei livelli sierici basali (pre-trattamento) alla sospensione, rappresenta un prezioso strumento per il trattamento dell’ipogonadismo, in particolare nell’anziano, dove l’eventuale comparsa di eventi avversi e/o complicanze (es. l’aumento dell’ antigene prostatico specifico, il PSA e/o il rilievo all’esplorazione rettale o all’ecografia di anomalie morfologiche a carico della prostata, ostruzioni acute delle vie urinarie) consente la rapida eliminazione dell’effetto androgenico esogeno, al contrario di quanto avviene con altre modalità di somministrazione (21-43). Controindicazioni alla terapia con testosterone includono ipertrofia prostatica, ginecomastia, insufficienza cardiaca congestizia ed eritrocitosi (2).
Va tenuto presente che nelle forme di ipogonadismo centrale isolato congenito da deficit isolato di gonadotropine sono stati descritti casi di ripresa spontanea del funzionamento dell’asse ipotalamo-ipofisi-gonadi con risoluzione del quadro clinico di ipogonadismo (17). Questo suggerisce pertanto la necessità di rivalutare nel tempo questi pazienti, mediante sospensione della terapia per un tempo adeguato e nuovo testing ormonale basale (44).
Bibliografia:
-
Peeyush Kumar, Nitish Kumar, Devendra Singh Thakur, and Ajay Patidar Male hypogonadism: Symptoms and treatment J Adv Pharm Technol Res. 2010 Jul-Sep; 1(3): 297–301.
- Basaria S Male hypogonadism. Lancet. 2014 Apr 5;383(9924):1250-63. doi: 10.1016/S0140-6736(13)61126-5. Epub 2013 Oct 10.1.
- A. Maestre de San Juan. Teratolagia: falta total de los nervios olfactorios con anosmia en un individuo en quien existia una atrofia congenita de los testiculos y miembro viril, El Siglo Medico, 1856, 211.
- G. De Morsier. Etudes sur les dysraphies cranioencephaliques, Arch Neurol Neurochir Psychiatr, 1954
- A. Ballabio e G. Camerino. The gene for X-linked Kallmann syndrome: a human neuronal migration defect, Curr Opin Genet Dev, 1992, 2, 417-421
- E. Rugarli e A. Ballabio. Kallmann Syndrome. From Genetics to neurobiology. JAMA, dicembre 1993
- Franco, B., Guioli, S., Pragliola, A., Incerti, B., Bardoni, B., Tonlorenzi, R., Carrozzo, R., Maestrini, E., Pieretti, M., Taillon-Miller, P., et al. (1991). A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 353, 529-536.
- Legouis, R., Hardelin, J.-P., Levilliers, J., Claverie, J.-M., Compain, S., Wunderle, V., Millasseau, P., Le Paslier, D., Cohen, D., Caterina, D., et al. (1991). The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell 67, 423-435.
- Ballabio, A., and Camerino, G. (1992). The gene for X-linked Kallmann syndrome: a human neuronal migration defect. Curr Opin Genet Dev 2, 417-421.
- Cadman SM, Kim SH, Hu Y, Gonzalez-Martinez D, Bouloux PM. Molecular pathogenesis of Kallmann’s syndrome. Horm Res 2007; 67(5):231-242.
- Franco B, Guioli S, Pragliola A et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 1991; 353(6344):529-536.
- Legouis R, Hardelin JP, Levilliers J et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell 1991; 67(2):423-435.
- Hardelin JP, Levilliers J, del C, I et al. X chromosome-linked Kallmann syndrome: stop mutations validate the candidate gene. Proc Natl Acad Sci U S A 1992; 89(17):8190-8194.
- F. Kallmann; W.A. Schoenfeld; S.E. Barrera. The genetic aspects of primary eunuchoidism, American Journal of Mental Deficiency, 1943-1944, 48, 203-236
- Quinton R, Duke VM, Robertson A et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol (Oxf) 2001; 55(2):163-174.
- Bonomi M, Libri DV, Guizzardi F et al. New understandings of the genetic basis of isolated idiopathic central hypogonadism. Asian J Androl 2012; 14(1):49-56.
- De RN, Young J, Misrahi M et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med 1997; 337(22):1597-1602.
- Dode C, Levilliers J, Dupont JM et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 2003; 33(4):463-465.
- De RN, Young J, Brailly-Tabard S, Misrahi M, Milgrom E, Schaison G. The same molecular defects of the gonadotropin-releasing hormone receptor determine a variable degree of hypogonadism in affected kindred. J Clin Endocrinol Metab 1999; 84(2):567-572.
- Sykiotis GP, Plummer L, Hughes VA et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U S A 2010; 107(34):15140-15144.
- Nachtigall LB, Boepple PA, Pralong FP, Crowley WF, Jr. Adult-onset idiopathic hypogonadotropic hypogonadism–a treatable form of male infertility. N Engl J Med 1997; 336(6):410-415.
- Katz DJ, Nabulsi O, Tal R, Mulhall JP.: Outcomes of clomiphene citrate treatment in young hypogonadal men. BJU Int. 2011 Nov 1.
- Bals-Pratsch M, Knuth UA, Yoon YD, Nieschlag E. Transdermal testosterone substitution therapy for male hypogonadism. Lancet 1986, 2: 943-6
- Behre HM, von Eckardstein S, Kliesch S, Nieschlag E. long term substation therapy of hypogonadal men with trans-scrotal testosterone over 7-10 years. Clin Endocrinol (Oxf) 50: 629-35
- Brocks DR, Meikle AW, Boike SC, Mazer NA, Zariffa N, Audet PR, Jorkasky DK. Pharmacokinetics of testosterone in hypogonadal men after transdermal delivery: influence of dose. J Clin Pharmacol 1996, 36: 732-9
- Cunningham GR, Cordero E, Thornby JI. Testosterone replacement with transdermal therapeutic system. Physiological serum testosterone and elevated dihydrotestosterone levels. JAMA 1989, 261: 2525-30
- Dobs AS, Meikle AW, Arver S, Sanders SW, Caramelli KE, Mazer NA. Pharmacokinetics, efficacy, and safety of a permeation-enhanced testosterone transdermal system in comparison with bi-weekly injections of testosterone enanthate for the treatment of hypogonadal men. J Clin Endocrinol Metab 1999, 84: 3469-78
- Findlay JC, Place Va, Snyder PJ. Transdermal delivery of testosterone. J Clin Endocrinol Metab 1987, 64:266-8
- Jordan WP, Atkinson LE, Lai C. Comparison of the skin irritation potential of two testosterone transdermal system: an investigational system and a marketed product. Clin Ther 1998, 20: 80-7
- Jordan WP. Allergy and topical irritation associated with transdermal testosterone administration: a comparison of scrotal and nonscrotal transdermal system. Am J Cont Dermat 1997, 8: 108-13
- Liverman CT, Blazer DG. Testosterone and Aging: Clinical Research Directions. Washington: DC: National Academies Press. 2004
- Meikle AW, Arver S, Dobs AS, Sanders SW, Rajaram L, Mazer N. Pharmacokinetics and metabolism of a permeation-enhanced testosterone transdermal system in hypogonadal men: influence of application site — a clinical research center study. J Clin Endocrinol Metab 1996, 81: 1832-40
- Nieschlag E, Swerdloff R, Behre HM, Gooren LJ, Kaufman JM, Legros JJ, Lunenfeld B, Morley JE, Schulman C, Wang C, Weidner W, Wu FC. Investigation, treatment and monitoring of late-onset hypogonadism in males: ISA, ISSAM, and EAU recommendations. Int J Androl, 2005, 28:125
- Parker S, Armitage M. experience with transdermal testosterone replacement therapy for hypogonadal men. Clin Endocrinol (Oxf) 1999, 50: 57-62
- Rolf C, Knie U, Lemmnitz G, Nieschlang E. interpersonal testosterone transfer after topical application of a newly developed testosterone gel preparation. Clin Endocrinol (Oxf). 2002, 56: 637-41
- Schaison G, Nahoni K, Couzinet B. Percutaneous dihydrotestosterone treatment. In: Nieschlag E, Behre HM eds. Testosterone: action, deficiency, substitution. Berlin: Springer Verlag. 1990, 155-64
- Steidle C, Schwartz S, Jacoby K, Sebree T, Smith T, Bachand R. North American AA2500 T Gel Study Group AA2500 testosterone gel normalizes androgen levels in aging males with improvements in body composition and sexual function. J Clin Endocrinol Metab 2003, 88: 2673-81
- Swerdloff RS, Wang C, Cunningham G, et al. Long-term pharmacokinetics of transdermal testosterone gel in hypogonadal men. J Clin Endocrinol Metab 2000, 85: 4500-10
- Wang C, Berman N, Longstreth JA, Chuapoco B, Hull L, Steiner B, Faulkner S, Dudley Re, Swerdloff RS. Pharmacokinetics of transdermal testosterone gel in hypogonadal men: application of gel at one site versus four sites: a general clinical research center study. J Clin Endocrinol Metab 2000, 85: 964-9
- Mc Nicholas TA, Dean JD, Mulder H, Carnegie C, Jones NA. A novel testosterone gel formulation normalizes androgen levels in hypogonadal men, with improvements in body composition and sexual function. BJU Int 2003, 91: 69-74
- Marbury T, Hamill E, Bachand R, Sebree T and Smith T. Evaluation of the pharmacokinetics profiles of the new testosterone topical gel formulation. Testim, compared to AndroGel. Biopharm Drug Dispos 2003, 24: 115-20
- Korenman SG, Viosca S, Garza D et al. Androgen therapy of hypogonadal men with transscrotal testosterone system. Am J Med 1987, 83:471-8
-
Bhasin S1, Cunningham GR, Hayes FJ, Matsumoto AM, Snyder PJ, Swerdloff RS, Montori VM: Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010 Jun;95(6):2536-59. doi: 10.1210/jc.2009-2354.
- Raivio T, Falardeau J, Dwyer A et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med 2007; 357(9):863-873.
1 commento
Hi there Dear, are you genuinely visiting this web
page regularly, if so then you will definitely obtain good knowledge.